1 Producción de penicilinasas: Fue Abraham, en 1940, el decubridor de estas enzimas inactivadoras de penicilinas y Kirby quien describió, en 1944, las características clínicas causadas por las bacterias que las producían. Ya a mediados de la década de los 50, se reportaba más del 50 % de las cepas de estafilococos hospitalarios resistentes a las penicilinas naturales por este mecanismo. Este fenómeno se fue extendiendo a diferentes especies bacterianas por distintos mecanismos de transmisión; cromosómicos, por plásmidos, tramposones y bacteriófagos.
Estas penicilinasas producidas por las bacterias grampositivas, son vertidas en el espacio extracelular donde inactivan a la penicilina antes de contactar a la bacteria; en el caso de las gramnegativas las enzimas son secretadas en el espacio periplásmico, inactivando el medicamento después de atravesar la pared bacteriana. Estas enzimas al ponerse en contacto con la penicilina rompen su anillo betalactámico transformándola en ácido penicilánico inactivo. Esta es hoy la principal forma de resistencia bacteriana a las penicilinas.
2 Cambios en las proteínas fijadoras de penicilinas: La aparición de genes mutantes en las bacterias, provoca sustituciones de alguna PFPs, lo que acarrea cambios en la afinidad por los antibióticos. Este fenómeno es el principal responsable de la creciente resistencia que vienen presentando ante los betalactámicos, cocos grampositivos (neumococos, estafilococos meticilina resistentes, enterococos) y otros, tales como, gonococo, H. influenzae, P. aeruginosa.
3 Ausencia en la activación de las enzimas autolíticas: Cuando ocurre este fenómeno, conocido como tolerancia, las concentraciones bactericidas mínimas; se tornan 32 veces mayores que las concentraciones inhibitorias mínimas; por tal razón las penicilinas se comportan como bacteriostáticos, el caso típico es el enterococo; puede presentarse también en estafilococos y estreptococos del grupo B.
4 Ausencia de pared y localización intracelular: Teniendo en cuenta su mecanismo de acción (inhibiendo la formación de la pared) y por su imposibilidad de penetración intracelular, toda bacteria con ausencia de pared o localización intracelular es resistente a las penicilinas: micoplasmas, clamidias, legionelas, brucelas, etc.
5 Ausencia de permeabilidad de la pared bacteriana: Este fenómeno está regido por la hidrofília, las cargas eléctricas y el tamaño molecular del antibiótico, debido a que la membrana externa de las bacterias gramnegativas, formada por fosfolípidos y lipopolisacáridos, es hidrófoba y posee unos canales acuosos formados por proteínas, denominados porinas, que atraviesan la membrana, y es por donde tienen que penetrar los antibióticos. En el caso de las pseudomonas, a pesar de contar con las 8 PFPs, solo las carboxipenicilinas y las ureidopenicilinas son capaces de atravesar su pared y llegar a la unión con las PFPs. Otro fenómeno relacionado con la permeabilidad, es la aparición de mutantes que provocan cambios en las porinas, trayendo consigo la imposibilidad de penetración del antibiótico, (ej: gonococo, E. coli).
Algunos parámetros farmacocinéticos de las penicilinas más usadas

Características generales de los principales tipos de penicilinas.
Bencilpenicilinas. La peniclina G resulta todavía una de las más activas frente a microorganismos gram (+) aerobios y anaerobios; son sensibles los estafilococos (no productores de penicilinasa), casi todos los estreptococos (pero no los enterococos), Neiserias gonorreae y meningitidis, Clostridium tetani, Corynebacterium diphteriae, Bacillus anthracis, Listeria monocytogenes, Treponema pallidum, Actinomyces rotesi, Leptospira, Streptobacillum moniliformis, Pasteurella multocida, Borrelia burgdorferi. Se distribuyen ampliamente a todo el organismo unidas a proteínas plasmáticas, principalmente a albúmina. Atraviesa el líquido cefalorraquídeo, también pasa al líquido pleural, sinovial, ascítico y pericárdico; al líquido amniótico y a la circulación fetal. No se metaboliza en el hígado, se eliminan por el riñón fundamentalmente en forma activa y en menor cuantía por bilis, leche materna y saliva. Semi desintegración corta (30 minutos), que aumenta en recién nacidos y en casos de insuficiencia renal o hepática.
Meticilina e isoxazolilpenicilinas. Espectro Estafilococco aureus productores de penicilinasa. Menos efectivas para los microorganismos sensibles a la penilina G. La meticilina es inactiva por vía oral, mientras las isoxazolil penicilinas si se pueden administrar por vía oral. La meticilina tiene menor afinidad por las proteínas plasmáticas que las penicilinas G, y las isoxazolil penicilinas, mayor afinidad. Excreción renal y hepática.
Aminopenicilinas. Tienen espectro similar a la penicilina G, pero además actúan contra gram (-) H. Influenzae, E. Coli, Samonella, Shigella y P. Mirabilis y vulgaris. Son destruidas por betalactamasas de microorganismos gram (+) y (-). Estables en medio ácido, por lo que se absorben por vía oral. La absorción del ampicillin es más estable y completa, por lo que la incidencia de diarreas es menor. Menos afinidad por las proteínas plasmáticas que la peniclina G. La ampicilina sufre circulación enterohepática y se excreta por la bilis. Excreción renal activa.
Carboxipenicilinas. Son activas frente a Pseudomonas aeroginosas, Proteus indol (+) resistentes a la ampicilina, Bacteroides fragilis. La carbenicilina es similar a la amoxacilina frente a H. influenzae, E. Coli, enterobacter y Samonella. La ticarcilina es de 2-4 veces más activa que la carbenicilina frente a P. aeroginosa, Enterobacterias y Neiserias. La carbenicilina y ticarcilina no son absorbidas por vía oral, pero la indanil carbenicilina si puede ser administrada por dicha vía. La unión a proteínas plasmáticas es de alrededor del 50% y la vida media de eliminación de cerca de 1 hora, demorándose hasta 2 horas en caso de insuficiencia renal. Se excretan por el riñón en forma activa en un 80% en 9 horas.
Ureidopenicilinas. La azlocilina y piperacilina son más activas que la carbeniclina y ticarcilina contra la P.aeroginosa. La piperacilina y mezlocilina son activas contra klebsiella, E. Coli, Serratia, Proteus, Enterobacter. No se absorben por vía oral adecuadamente. La vida media de eliminación es de una hora con función renal normal y hasta cinco si existe insuficiencia renal. Excreción biliar alta.
Amidinopenicilina. Mecillinám. Espectro: E.coli, Proteus, mirabilis, Klebsiella, Enterobacter, Salmonella, Shigella, Serratia. La vida media de eliminación es de una hora y se elimina por vía renal sin modificar en un 70%.
Efectos indeseables de las penicilinas.
Hipersensibilidad: fiebre, asma, púrpura trombocitopénica, anemia hemolítica, neutropenia, pancitopenia, vasculitis, urticaria y shock anafiláctico. La incidencia de estas manifestaciones llega a un 5–10 % de los pacientes tratados.
Sistema Nervioso Central: convulsiones, hiperreflexia y coma por efecto irritativo de las altas concentraciones de penicilina G en el líquido cefalorraquídeo.
Hiperpotasemia, sobre todo en pacientes con insuficiencia renal por la propia penicilina G potásica.
Hipopotasemia y sangramiento por carbenicilina.
Descompensar una insuficiencia cardiaca: la penicilina G y otras penicilinas sódicas (carbenicilina).
Nefritis intersticial: penicilinas semisintéticas (meticilina y dicloxacilina).
Trastornos gastrointestinales: náuseas, vómitos y diarreas las de uso oral.
La administración IM puede dar lugar a abscesos y la EV a flebitis.
Fenómenos de suprainfección graves por microorganismos productores de penicilinasa.
Interacciones e incompatibilidades.
Actividad sinérgica con muchas otras drogas antimicrobianas tales como: aminoglucósido, cloranfenicol, sulfonamidas, fosfomicina, etc. Que pueden aumentar su efecto.
Los salicilatos (ASA incluida) son capaces de desplazarlas de su unión a las proteínas plasmáticas y aumentar la fracción libre de las mismas.
La fenilbutazona, la sulfinpirazona y el probenecid al competir con la excreción tubular renal de las mismas También aumentan su efecto.
Los antagonismos se pueden presentar en: Fase farmacéutica, las penicilinas que se emplean por vía parenteral se inactivan a pH inferiores a 8, por lo que no se debe administrar con soluciones ácidas ni alcalinas. La penicilina G no se debe administrar conjuntamente con soluciones de lactato o bicarbonato, ni conjuntamente con anfetericina B, cefalotina sódica, clorpromacina, heparina sódica, histamina, novobiocina sódica, pentobarbital sódico, prometacina ni tetraciclinas.
La meticilina no se debe administrar con ClNa 0.9 %, solución de dextrosa, de lactato o bicarbonato. Tampoco conjuntamente con aminoglucósidos.
La ampicilina sódica no debe administrarse conjuntamente con drogas que varíen su pH y afecten la estabilidad como: atropina, cloranfenicol, tetraciclina, noradrenalina, novobiocina, pentobarbital y fenobarbital sódico, prometacina, sulfisoxazol, tiopental sódico y vitaminas del complejo B.
La cabenicilina sódica no se recomienda su administración conjunta con: anfotericina B, cloranfenicol, aminoglucósidos, tetraciclinas y vitaminas del complejo B y C.
En la fase farmacocinética, la ampicilina reduce la eficacia de los contraceptivos orales por inducción enzimática o mala absorción intestinal.
Usos clínicos.
Aunque nuevos fármacos permiten al médico disponer de diversas opciones de tratamiento, las penicilinas han sido sustancias de gran éxito: son rápidamente bactericidas, bien toleradas y eficaces, tanto por vía oral como parenteral.
Otitis y sinusitis (durante la infancia): ampicilina o amoxacilina (Hemophilus influenzae). En infecciones crónicas: penicilinas antipseudomonas y antiestafilocócicas.
Faringitis: penicilina G o la penicilina V (Streptococcus pyogenes).
Epiglotitis: ampicilina o amoxacilina (Hemophilus influenzae) asociado al cloranfenicol.
Bronquitis: ampicilina o amoxacilina (Neumococo, Branhamella catarrhalis).
Neumonía: lactantes y niños menores de 4 años: amoxacilina con clavulanico (Hemophilus productor de penicilinasa). En niños mayores y adultos: penicilina G (neumococo). En ancianos, diabéticos, alcohólicos y pacientes con fibrosis quística: penicilina antipseudomonas.
Meningitis: neonato: penicilina antispeudomona. Adulto: penicilina G y ampicilina.
Infecciones de vías urinarias: ampicilina, amoxacilina, penicilina antipseudomona.
Infecciones intra abdominales: penicilina antipseudomona.
Infecciones biliares: ampicilina, amoxacilina.
Infecciones venéreas: penicilina G (Sífilis).
Infecciones entéricas: ampicilina.
Infecciones cutáneas y de tejidos blandos: penicilinas antipseudomona y antiestafilococcica.
Infecciones de huesos y articulación: penicilinas antipseudomona y antiestafilococcica.
Bacteremia: penicilinas antipseudomona y antiestafilococcica.
Endocarditis: penicilina G (válvulas naturales); penicilina antiestafilococcica (prótesis)




B. Cefalosporinas
Historia.
La primera cefalosporina, conocida como C, la obtuvo el Dr. Guisseppe Brotzo, en 1945, de un hongo (Cephalosporium Acremonium) aislado de aguas albañales, en la costa de Cerdeña. En 1960 es identificado el núcleo de las cefalosporinas, el ácido 7 amino-cefalosporánico, locuaz favorecio el desarrollo de lasa cfalosporinas semisintéticas, apareciendo en 1964 la cefalotina como primer representante del grupo, más tarde van apareciendo el resto de las cefalosporinas de 1ra generación.
Años más tarde, en la década del 70, surgen las cefalosporinas de 2da generación, incluidas en este grupo las cefamicinas (cefoxitina, cefotetan, cefmetazol), obtenidas a partir de 8 especies de streptomyces, cuya diferencia química con las cefalosporinas es la presencia de un grupo alfa-metoxi en la posición 7.
A principios de la década del 80, entran en el mercado las cefalosporinas de 3ra generación, cuyo primer componente fue el cefotaxime. Por último aparecen en el mercado, en un momento de desesperación infecciosa, provocado por el número cada vez más creciente de cepas bacterianas multirresistentes, las cefalosporinas de 4ta generación, encabezadas por el cefepime y el cefpirome.
Clasificacion.
Se clasifican en "generaciones" de acuerdo al orden de aparición, las características farmacocinéticas y la actividad antimicrobiana.
1RA Generación (1964- 1969)
Oral: Cefalexina, Cefadroxil, Cefadrina, Cefaloglicina, Cefatrizina, Cefsumida, Cefedeclor, Cefroxadina, Pivcefalexina
Parenteral: Cefalotina, Cefaloridina, Cefazolina, Cefapirina, Cefacitrile, Cefalixina, Cefadrina, Cefanona, Cefazedona, Ceftezol, Cefazaflur, Cefaloglicina
2da Generación (1970-79)
Oral: Cefuroxime, Cefaclor, Cefatrizina, Cefprozilo, Loracarbef, Ceftidoren.
Parenteral: Cefamandol, Cefoxitina, Cefotetan, Cefuroxima, Cefonicida, Ceforanida, Ceftemazol, Cefbuperazona, Cefminox, Cefotiam, Flomoxef
3ra Generación (1980- 89)
Oral: Cefixima, Cefpodoxima, Cefnidin, Cefetamet, Ceftibuten, Ceftetam, Cefditoren
Parenteral: Cefotaxime, Moxalactam, Ceftizoxima, Ceftriaxona, Cefmenoxima, Cefodizina, Cefpiramida, Cefozonan, Latamoxef, Flumoxef
Actividad Antipseudomona: Cefoperazona, Ceftazidima, Cefsulodina
4ta Generación (1995- 97)
Parenteral: Cefpirome, Cefepima, Cefaclidina, Cefelidina, Cefoselis, Cefozopran, Cefempidoma, Ceftiofur, Ceftobiprole.
Química.
Todas las cefalosporinas contienen un anillo betalactámico de cuatro miembros fijos a un anillo de dihidrotiacina de seis miembros, llamado ácido 7 amino-cefalosporánico (ver estrutura de las cefalosporinas anteriomente). Las sustituciones en las cadenas laterales (posición 7) se relacionan con el espectro antimicrobiano y la estabilidad intrínseca a las betalactamasa y los cambios en la posición 3 desempeñan un papel importante en la farmacocinética y en la toxicidad. Ej: Ceftriaxona tiene semidesintegración prolongada. Mediante otros cambios en la estructura básica se han obtenido compuestos muy similares estructuralmente, aunque no pertenecen a las cefalosporinas: Cefamicinas: Cefotetan y cefoxitina. Oxabetalactámico: Moxalactam
Mecanismo de acción.
Como las penicilinas inhiben una serie de enzimas que catalizan pasos importantes en la formación de la pared celular bacteriana. De igual manera que la peniclinas, los distintos componentes de la familia de las cefalosporinas, tienen afinidades independientes por determinados número de PFPs, no obstante bloquean preferentemente Ia y Ib, con escasa acción sobre 4,5 y 6.
Caracteristicas generales de los principales grupos:
1ra Generación. Todas las pertenecientes a este grupo poseen similar espectro antimicrobiano, o sea son eficaces contra: Todos los cocos gram (+), excepto los Streptococcus faecalis y staphylococcus aureus resistentes a la meticilina. Aerobios gram (-) como E. Coli, Klebsiella peumoniae, P. mirabilis, Clostridium, Listeria monocytogenes, salmonella spp, Shigella spp. La mayoría de los anaeróbicos excepto el B. Fragilis. Todos atraviesan la barrera hematoencefálica, aunque no alcanzan concentraciones terapéuticas elevadas. Sin embargo se distribuyen bien en fluidos pleural, pericardio y sinovial y se eliminan completamente por el riñón. Son nefrotóxicas.
2da Generación. Las de este grupo tienen un espectro mayor que incluyen: Gram (-) resistentes a las de la primera generación: Proteus indol (+), Enterobacter y H. influenzae. –B. Fragilis el cefotetan y la cefoxitina. No atraviesan la barrera hematoencefálica, excepto la cefuroxima; son inefectivas contra las Pseudomonas y Enterococos y son relativamente ototóxicas.
3ra Generación. Esta generación tiene un amplio espectro, sobre todo gram (-) como: Enterobacter, Serratia spp, Citrobacter, H. influenzae, N. Gonorrhoeae y los anaeróbicos. Contra la P. aeroginosa la ceftazidima, cefoperazona y cefsulodina. Todas alcanzan concentraciones bactericidas en el líquido cefalorraquídeo. Son las que presentan más larga vida media de eliminación, por lo que mantienen un alto grado de actividad incluso a bajas concentraciones séricas. No poseen toxicidad significativa, ni cuando se utilicen a altas dosis.
4ta Generación. Las nuevas cefalosporinas poseen una estructura química que las hace tener una buena penetración a través de la membrana celular más externa de las bacterias y poca afinidad por las betalactamasas tipo 1, lo que reduce su degradación enzimática en comparación con otras cefalosporinas. Tienen un espectro de actividad más amplio, que incluye: cepas de Enterobacteriaceae resistentes a la ceftazidima. Microorganismos gram (+) importantes como el S. Aureus (no meticilino-resistente) y el Streptococcus pneumoniae penicilino resistente. Gram (-) como la P. Aeroginosa. Se administran por vía parenteral, cruzan la barrera hematoencefálica de manera excelente y se excretan por el riñón. Tienen baja toxicidad y se toleran localmente.
Resistencia bacteriana.
La principal causa de resistencia es la producción de betalactamasa o en este caso cefalosporinasa, la cual puede ser inducida por mutaciones cromosómicas o mediadas por plasmidas. Las nuevas (3ra generación) son sumamente resistentes a la hidrólisis de las enzimas de los gramnegativos y sólo cefoxitina, cefotetan y moxalactan muestran estabilidad contra las producidas por bacteroides fragilis
En los últimos años se ha visto un incremento de betalactamasas de espectro extendido (BLEE) derivadas de las enzimas TEM o SHI, las cuales provocan resistencia a las oxiiminocefaslosporinas (ceftriaxona, ceftazidima, cefotaxima) de múltiples enterobacterias, siendo mas frecuente: E. coli, Klebsiella, Proteus, Providencia y Enterobacter. Recientemente se ha reportado una familia de plásmidos de estas betalactamasas de amplio espectro, con preferencia por el cefotaxime. También el uso repetitivo de estas cefalosporinas provoca una desinhibición del Gen para la producción de la enzima AmpC inducible, presente fundamentalmente en: Enterobacter, Citrobacter, Pseudomona, E. coli y K. pneumoniae.
En 1990, fueron descubiertas betalactamasas resistentes al ácido clavulánico y sulbactam, las cuales permanecen sensibles al tazobactam. Son variantes TEM 1 y TEM 2, de las que se han descrito alrededor de 17 tipos.
Otros mecanismos de resistencia ante las cefalosporinas son: la disminución de la permeabilidad bacteriana ocasionada por mutaciones en los genes que codifican las porinas (OMP) y las alteraciones en las PFPs.
Algunos parámetros farmacocinéticos de las cefalosporinas más usadas

Reacciones adversa.
Producen pocas reacciones, entre las más importante tenemos:
Reacciones de hipersensibilidad: Fiebre, rash, enfermedad del suero, eosinofilia, anafilaxia, broncoespasmo y urticaria (de un 5-10 % de los pacientes alérgicos a las penicilinas pueden manifestar reactividad cruzada.
Reacciones locales: Dolor en el sitio de inyección IM y tromboflebitis por infusión EV.
Efectos similares a los producidos por el disulfiran (cefoperazona, cefamandol y moxalactan). Los efectos sistémicos son infrecuentes y pueden incluir:
Hematológicas: Test de Coombs positivo, anemia hemolítica, trombocitopenia y leucopenia reversibles y granulocitopenia por cefalotina.
Nefrotoxicidad (cefaloridina) cuando se usan más de 4 g/día, exista una enfermedad renal previa, en pacientes mayores de 60 años o con la administración simultánea de medicamentos nefrotóxicos como furosemida, ácido etacrínico y aminoglucósidos.
Hepatotoxicidad: hipotrombinemia (responde al tratamiento con vitamina K) y elevaciones transitorias de las enzimas hepáticas.
Trastornos gastrointestinales: náuseas, vómitos, diarreas, cólicos abdominales y colitis pseudomembranosa.
Interacciones e imcompatibilidades.
En la fase farmacéutica son incompatibles con compuestos de alto peso molecular y con metales alcalinoterreos. Además no deben adicionarse a soluciones que no tengan un pH entre 4 y 7, por lo que no deben mezclarse con Glutamato de calcio, metilprednisolona, aminofilina, eritromicina, tiopental, kanamicina, pentobarbital, tetraciclina, difenilhidantoína, penicilina G sódica o potásica, fenobarbital, polimixina B, sulfisoxazol ni con aminoglucósidos. En la fase farmacodinámica se han reportado antagonismos con: rifampicina, tetraciclina, cloranfenicol, ampicilina y carbenicilina. La cefaloridina o cefalotina cuando se administra conjuntamente con ácido etacrínico, furosemida o aminoglucósidos, aumenta la nefrototixicidad de ambos grupos de drogas. El probenecid y la sulfinpirazona inhiben su excreción renal, por lo que pueden aumentar sus concentraciones séricas.
Uso clínico:
Infecciones estafilocócicas (1ra Generación Cefazolina).
Profilaxis quirúrgica: 1ra Gen. Cefazolina; cirugía del tórax, abdomen y ortopedia: 2da Gen. Cefoxitina cirugía colorrectal.
Infecciones de vías urinarias: cefalosporinas orales: 1ra Gen. Cefalexina: 2da Gen. Cefaclor: 3ra Gen. Cefixime.
Infecciones óseas o articulares: Osteomielitis estafilocócica: 1ra Gen. Cefazolina. Artritis séptica 3ra Gen. Ceftazidima, Ceftriaxona.
Infecciones mixtas abdominal o pélvica: 2da Gen. Cefoxitina o cefotetan, 3ra Gen. Ceftriaxona.
Infecciones respiratorias: 2daGen. Cefaclor: sinusitis bacteriana, otitis media y bronquitis.2da Gen. Cefuroxima en neumonía por H. Influenzae, neumococo, estafilococo o klebsiella. 2da Gen. Cefoxitina en neumonía por aspiración (mixta). 3ra Gen. Ceftazidima en neumonía por pseudomona.
Infecciones por bacilos gram (-) entericos. -3ra Gen. Cefotaxima, ceftizoxima o ceftriaxona.
Meningitis en adultos y niños. 3ra Gen. Cefotaxima, ceftriaxona, ceftazidima. En recién nacidos: Cefotaxima + Ampicillin. Si pseudomona, ceftaxidima.
Septicemia y neutropénicos febriles: 3ra Gen. (Ceftaxidima) + Aminoglucósidos.
Enfermedades de transmisión sexual: Ceftriaxona IM para gonorrea y chancro blando. Enfermedad de Lyme. Ceftriaxona EV.
Presentación de algunas cefalosporinas




C. Carbapenémicos
Historia:
A finales de la década del 70, durante el análisis rutinario de microorganismos de la tierra en busca de nuevos inhibidores de la síntesis de los peptidoglicanos, fueron descubiertos los carbapenémicos. Esto ocurrió a partir de una nueva especie de Streptomyces, denominada Streptomyces cattleya, por la similitud de la pigmentación de sus esporas con la orquídea del mismo nombre. La estructura de la tienamicina, primera de este grupo, fue descubierta por Albert–Schonberg y col. en 1978, y la misma guarda notable semejanza con los betalactámicos corrientes. Por su gran inestabilidad fisicoquímica, este producto no pudo ser utilizado clínicamente. A través de los estudios de Leanza y col. se obtuvo una molécula más estable, sintetizándose el N-formidoiltienamicina, conocido como imipenem, congénere sintético de la tienamicina natural.
La tienamicina es inactivada, por una enzima renal, producida por la células del epitelio del túbulo proximal, una dipeptidasa, denominada deshidropeptidasa I, la cual actúa como una verdadera betalactamasa, dando lugar a un metabolito, el cual provoca daño renal por necrosis tubular aguda. Esta enzima no posee este tipo de acción sobre el resto de los betalactámicos. Los investigadores se dieron a la tarea de buscar una sustancia que bloqueara la acción de esta enzima renal, de esta investigación surgió la cilastatina, la cual posee una farmacocinética similar a la tienamicina y no actúa sobre otras zincmetalopeptidasas humanas del riñón, páncreas y pulmón (enzima convertidora de angiotensina).
Posteriormente apareció otro componente sintético de esta familia, el meropenem, con mayor resistencia a las betalactamasas que el imipenem, no siendo degradado por la deshidropeptidasa renal, por lo que no necesita ser combinado con la cilastatina. Este fue seguido por la aparición en el mercado de otros carbapenémicos y sulfopenémicos como; biapenem, panipenem, faropenem, sulopenem, ritipenem, sanfetrinem y ertapenem, este último aprobado por la FDA en 2001.
Clasificacion carbapenemos
Oral: Faropenem, Ritipenem, Sanfetrinem, Tebipenem.
Parenteral: Imipenem, Meropenem, Panipenem, Biapenem, Ritipenem, Sanfetrinem, Sudopenem, Lenapenem, Ertapenem, Doripenem.
Estructura química:
El imipenem es un derivado N-formidoil de la tienamicina y forma parte del grupo carbapenémico, pertenciente a la familia de los betalactámicos. Posse como ellos un anillo betalactámico central, con determinados cambios estructurales, siendo el más importante, la sustitución del átomo de azufre central por un grupo metilo, lo que aumenta su reactividad con las proteínas de la pared bacteriana, proporcinándole un potente efecto bactericidad. Otro cambio estructural, que le da su excepcional estabilidad frente a las betalactamasas, es la posición trans de su cadena lateral hidroxietílica unida al anillo central. Tanto el imipenem como el panipenem son inactivados por la deshidropeptidasa renal I, por tal motivo el imipenem está asociado a la cilastatina sódica (proporción de 1:1) un inhibidor de dicha enzima y el panipenem al betamepram (proporción de 1:1) un inhibidor del transporte de la enzima renal. El meropenem, biapenem y faropenem, son resistente a dihca enzima, por lo que no necesitan su unión a la cilastatina.
Mecanismo de acción.
Es semejante al de los betalactámicos, se une con las proteínas ligadoras de penicilinas (PBPs), interfiriendo en la síntesis de la pared bacteriana y culmina en la muerte del microorganismo. En orden decreciente de afinidad, el imipenem se une a los tipos 2, 1 y 3 de proteínas ligadoras de penicilina. El espectro extraordinariamente amplio de actividad se debe a su afinidad por PBPs críticas de muy diversos microorganismos. Además, al parecer, entra con facilidad en la pared bacteriana por su pequeño tamaño y estado de zwitterion, lo cual permite la fácil penetración a sus blancos posibles. El Imipenem posee extaordinaria estabilidad contra el ataque de las betalactamas, por la configuración trans, poco común de su cadena lateral de hidroxietilo. Tienen efecto postantibiótico contra Enterobacterias y P.aeroginosa y ello significa que después que la concentración del fármaco ha descendido por debajo de los niveles inhibidores, las bacterias que no han sido destruidas no reanudan su proliferación durante 2-4 horas.
Espectro de acción
Considerado hoy entre los antimicrobianos más potentes, su amplísimo espectro se extiende a bacterias gramnegativas, grampositivas, aerobios y anaerobios, por tal motivo mencionaremos las especies bacterianas que no son sensibles ante este grupo: Staphylococcus aureus y epidermidis meticilina resistente (SMR), enterococo vancomicina resistente (EVR), S.faecium, C. difficile, Corynebacterium JK, X. maltophila, X. cepacea, Flavobacterium, Chlamydia, Rickettsia, Legionella y M. fortuitum.
El meropenem al igual que el biapenem, tiene una acción más potente que el Imipenem sobre las bacterias gramnegativas (incluyendo la Pseudomona aeruginosa) y anaerobias, siendo menor sobre las grampositivas. El panipenem es el de mayor efectividad sobre las pseudomonas. El faropenem, no tiene acción sobre pseudomonas y Acinetobacter.
Mecanismos de resistencia
Los niveles de resistencia ante el imipenem son bajos, a pesar de su alta estabilidad ante las betalactamasas; ha sido descrita la resistencia de algunas cepas de B. fragilis, X. hydrophila, y P. aeruginosa por producción de batalactamasas cromosómicas. También ha aparecido la resistencia de cepas de P. aeruginosa en el curso de tratamientos, debido a cambios en sus porinas o en las PFP2.
El aumento del uso de los carbapenémicos en la década del 90, trajo aparejado el surgimiento de una serie de carbapenemasas, las cuales se han incrementado en los últimos años. La mayoría son metaloenzimas que son inhibidas por EDTA y se han descrito en B. fragilis, pseudomonas, aeromonas, L. gormanii, F. odoratum, B. Cereus.
Algunos parámetros farmacocinéticos de las carbapenemos más usadas

Efectos adversos
Como el resto de los betalactámicos su toxicidad es baja, pudiendo aparecer reacciones alérgicas caracterizadas por erupción cutánea, prurito, fiebre y urticaria.
Presenta hipersensibilidad cruzada con el resto de los betalactámicos por lo que no deben ser utilizados en caso de antecedentes de reacciones graves a los mismos.
Otros efectos menos frecuentes son: náuseas, vómitos, diarreas, colitis pseudomembranosa, leucopenia, trombocitopenia, anemia, prueba de Coombs positiva, elevación de transaminasas, bilirrubina y fosfatasa alcalina, insuficiencia renal aguda, mioclonía, trastornos confusionales y convulsiones, este último uno de sus efectos mas temidos sobre todo en niños, presentándose con menor frecuencia con el meropenem, por lo que es preferido para el uso pediátrico.
Caracteristicas generales de los principales medicamentos
Imipenem. Es un derivado del Streptomices cattleyar con un rango amplísimo de actividad antimicrobiana, es resistente a las betalactamasas de los gramnegativos y grampositivos, en particular aquéllas mediadas por plásmidos. La concentración sérica pico es de 21-58 Fg/mL, la vida media de eliminación sérica es de una hora y el 20 % de la droga se une a las proteínas del plasma. Se excreta por los riñones del 70-80 %, aunque no se observan concentraciones urinarias elevadas, ya que la droga se hidroliza en el túbulo proximal por la enzima dehidropeptidasa y produce metabolitos nefrotóxicos.
Complicaciones y efectos indeseables.
El imipenem ha sido señalado por la alta incidencia de convulsiones asociadas a su uso (más del 33 %) incluso en pacientes sin antecedentes, esto ha sido más frecuente en pacientes con VIH positivo. Otras complicaciones de su uso son la insuficiencia renal leve y elevación de las enzimas hepáticas. Reacciones locales: eritema, dolor e induración, tromboflebitis. Reacciones alérgicas y cutáneas: erupción cutánea, prurito, urticaria, eritema multiforme, edema angioneurótico, fiebre, reaciones anafilácticas. TGI: náuseas, vómitos, diarreas, manchas en los dientes y colitis pseudomembranosa. Hematológicas: eosinifilia, leucopenia, neutropenia, trombocitopenia.
Contraindicaciones.
Hipersensiblidad a cualquiera de los componentes de este producto.
Debido a que su diluente contiene clorhidrato de lidocaina (TIEMAN I.M), está contraindicado en pacientes con hipersensibilidad a los anestésicos locales de tipo amida.
Interacciones medicamentosas.
Se han observado convulsiones generalizadas en pacientes que recibieron ganciclovir y TIENAM EV. Estos dos medicamentos no se deben utilizar al mismo tiempo, a menos que los beneficios potenciales sean mayores que los riesgos.
Meropenem. Novedoso y potente antibiótico de amplio espectro, cuya eficacia ha sido probada en varios estudios. Es tolerado muy bien por el niño y el adulto, incluso en altas dosis, con muy pocos efectos secundarios como náuseas y vómitos; la administración endovenosa de hasta 6 g diarios se utiliza en los casos de meningitis en el adulto. Es estable ante la dehidropeptidasa renal humana y, a diferencia del imipenem, no necesita la administración conjunta de un inhibidor de esta enzima como la cilastatina; además, tiene una rápida acción bactericida de amplio espectro que cubre a gram (+), gram (-), aerobios y anaerobios, gérmenes resistentes a la tercera generación de cefalosporinas, y se utiliza por vía endovenosa e intramuscular con buenos resultados.
Ertapenem es otro antibiótico para el tratamiento parenteral de infecciones graves por patógenos gramnegativos o grampositivos, incluidos los anaerobios. A diferencia de los preparados de este grupo disponibles hasta la fecha (imipenem y meropenem), el ertapenem puede administrarse una vez al día, por vía intravenosa, a una dosis de 1,0 g. Gracias a su semivida más prolongada Al final de la infusión de 30 minutos de 1 g de ertapenem, las concentraciones plasmáticas medias en el adulto sano se situaron en 155 mg/l; al cabo de 12 horas, habían descendido a 9 mg/l y al cabo de 24 horas, a 1 mg/l. La semivida de eliminación es de aproximadamente cuatro horas, con lo que es claramente más prolongada que la de los restantes carbapenemos conocidos hasta la fecha. La fijación de la molécula a proteínas es relativamente elevada (92 a 95%); el volumen de distribución se cifra en 8 litros. La eliminación se efectúa principalmente por vía renal; en la orina, se observa en igual medida el fármaco inalterado o en forma de metabolito. La metabolización consiste en una hidrólisis del anillo betalactámico mediada por la dehidropeptidasa. En comparación con adultos sanos, los pacientes con una insuficiencia renal leve (aclaramiento de la creatinina: 31 – 59 ml/min/1,73 m2) presentan valores ABC de aproximadamente 1,5 a 1,8 veces superiores; esta alteración de la cinética no requiere de un ajuste de la dosis. Hasta la fecha, no se dispone de suficientes datos que permitan una recomendación sobre las dosis en los pacientes con insuficiencia renal avanzada. Esto también es aplicable a pacientes sometidos a hemodiálisis y a pacientes con insuficiencia hepática. En el ensayo clínico, demostró tener una buena tolerancia; por el momento, no se pueden dar datos definitivos sobre posibles efectos adversos raros. Es necesario adquirir una mayor experiencia con el ertapenem para poder definir mejor el lugar terapéutico que ocupará.
Doripenem fue lanzado por el Lab. Shionogi Co. de Japón bajo la marca registrada "Finibax" en 2005. Es particularmente activo contra la bacteria Pseudomonas aeruginosa. La Comisión Europea ha autorizado la comercialización en toda la Unión Europea el 25 de julio de 2008. Antibacteriano de amplio espectro, inyectable, , con la ventaja sobre el resto de la familia de carbapenems de ser más estable una vez diluido y poder mantener la infusión del antibiótico hasta 1 hora y en el caso de neumonía asociada a ventilación mecánica, se admiten infusiones hasta 4 horas. Las indicaciones para las que está aprobado son: Neumonía nosocomial, incluida la neumonía asociada a ventilación mecánica. Infección intraabdominal complicada. Infecciones urinarias complicadas, incluida pielonefritis.
Usos de los carbapenémicos:
Infecciones nosocomiales. Por microorganismos con resistencia a múltiples antibióticos, particularmente citrobacter freundi y especie de enterobacter.
Sepsis intraabdominal. El meropenem y el imipenem han sido utilizados como monoterapia en la sepsis intraabdominal con muy buenos resultados, y existen estudios que los colocan al mismo nivel que la combinación de cefotaxime y metronidazol y la clindamicina con tobramicina.
Sepsis del sistema nervioso central. Son altamente efectivos in vitro contra los microorganismos patógenos causantes de meningitis. Ambos, el imipenem y el meropenem penetran el líquido cefalorraquídeo (LCR) con las meninges inflamadas. El meropenem en particular es similar a la cefotaxima en la terapéutica de estas afecciones, no causa convulsiones y es efectivo en la meningitis por P. aeruginosa multirresistentes.
Sepsis urinaria. El meropenem y el imipenen han sido utilizados en el tratamiento de la sepsis urinaria complicada con muy buenos resultados, sus dosis respectivas son de 500 mg c/8h y 500 mg c/6h EV.
Paciente neutropénico. El uso del imipenem y el meropenem en el paciente inmunocomprometido ha sido avalado por varios autores, y ha sido comparado su empleo con la combinación de ceftazidima y amikacina, con iguales resultados que esta asociación y con menos complicaciones renales.
Sepsis ginecológica. El meropenem, por su amplia cobertura contra grampositivos, gramnegativos y anaerobios son medicamentos de elección en las sepsis obstétricas y ginecológicas complicadas; en estudios comparativos se ha demostrado la superioridad de la monoterapia carbapenémica sobre combinaciones de cefalosporinas y aminoglucósidos.
Sepsis respiratoria. Se han recomendado en el tratamiento de la sepsis respiratoria, incluso en aquella concomitante con una enfermedad pulmonar obstructiva crónica (EPOC), por su efecto bactericida frente a H. influenzae, S. pneumoniae, S. aureus, P. aeruginosa, M. catarrhalis, Klebsiella, Proteus, Enterobacter spp y otras bacterias grampositivas; especialmente el meropenem tiene una eficacia clínica similar a la ceftazidima y a la amikacina y tiene la ventaja de provocar menos efectos colaterales que el imipenem.
Otras sepsis graves. También han sido usados con magníficos resultados en las infecciones de la piel; el meropenem penetra en el tejido de las válvulas cardíacas, y es por lo tanto útil en la endocarditis infecciosa (EI). En estudios experimentales se ha demostrado su eficacia también en la endoftalmitis staphyloccócica en el conejo, se obtienen iguales resultados que con quinolonas y vancomicina.

D. Monobactámicos
Historia
A mitad de la década del 70, los laboratorios Squibb para investigaciones médicas de Pricenton, New Jersey, abandonaron las investigaciones de antibióticos producidos por hongos Streptomyces y se dieron a la tarea de estudiar otros microorganismos. En esta actividad descubrieron los monobactámicos, sustancias antibióticas que poseen un núcleo betalactámico, semejante a las betalactamasas, pero careciendo de núcleo secundario condensado con aquel. Estos productos tienen su origen en bacterias grampositivas (Nocardia) y gramnegativas que habitan en el suelo (Gluconobacter, Acetobacter, Agrobacterium, Chomobacterium, Pseudomona).
La primera sustancia de este grupo fue hallada en una muestra de tierra de un pinar al sur de New Jersey, donde se aisló una cepa de Chomobacterium violaceum, la cual producía una molécula betalactámica, con actividad antibiótica hasta ese momento desconocida. Estas moléculas naturales exhibían escasas propiedades antibacterianas.
Más tarde, usando su núcleo monocíclico (ácido 3- aminomonobactámico) sintetizado químicamente, adicionándole diversos radicales, se han obtenido más de un millar de derivados semisintéticos y sintéticos con disímiles propiedades biológicas. Dentro de este grupo, por sus características farmacocinéticas, fue seleccionado el aztreonam como el primer monobactámico para la utilización clínica en la década del 80, su actividad se asemeja a la de los aminoglucósidos y es muy activo frente al Gonococo y el H. influenzae. Tiempo después aparecieron otros productos dentro de esta familia: carumonam, tigemonam, oximonam, gloximonam, pirazonam, que ya comienzan a ser introducidos en el mercado por su actividad esencialmente contra gérmenes gramnegativos.
Clasificacion monobactámicos:
Oral: Tigemonan, Gloximonan, Monosulfactam.
Parenteral: Aztreonam, Carumonan, Pirazonam, Oximonam.
Estructura química:
Su estructura con un sólo anillo central (monobactámico),
echó por tierra la creencia de que era necesario un segundo anillo condensado
con el principal para tener actividad antibacteriana. Estos productos poseen
un grupo sulfónico en la posición I, responsables por la activación
del núcleo betalactámico y su efectividad contra las pseudomonas,
teniendo además un radical acílico en la posición 3 beta
que le confiere excelente activiadad contra bacterias gram (-) y un grupo alfa
metoxi en posición 4 que le da su gran estabilidad contra betalactamasa.
Mecanismo de acción.
Los monobactámicos interfieren en la síntesis de la pared bacteriana,
se liga preferentemente a la proteína 3 ligadora de penicilina (PBP3)
de gramnegativos aeróbicos. Por interactuar pobremente con las PFPs 1a,
1b y 2 su acción sobre bacterias grampositivas y anaerobios resulta igualmente
pobre. Una de las ventajas sobre los demás betalactámicos es su
capacidad de penetrar dentro de mácrofagos y granulocitos, consiguiendo
buenas concentraciones intracelulares.
Espectro antimicrobiano.
De forma general tiene excelente acción sobre las bacterias aerobias gram (-). Especialmente contra el Gonococo y el H. influenzae, Entorobacter y Pseudomonas. Su espectro de acción alcanza también a Salmonella, Shigella, Proteus, Meningococo, Hafnia alvei, Serratia, E. coli, Providencia, Citrobacter freundii, E. cloacae, Klebsiella y Morganella. Los monobátamicos orales (tigemonam, gloximonam) no tienen actividad frente a pseudomona, Acinetobacter, Bordetella y Mroaxella. El pirazonam presenta gran potencia frente a pseudomonas y Acinetobacter, quizás más potente que la ceftazidima.
Mecanismo de resistencia:
La betalactamasa cromosómica, como las de el E. cloacae, son los mayores responsables de los fallos terapéuticos. Sin embargo los bacteroides aunque no son sensibles, sus betalactamasas pueden ser inhibidas por estos compuestos; este efecto sumado a su importante espectro sobre los gramnegativos, justifica la asociación de monobactámicos y antianaeróbicos en las sepsis mixtas (abdominales, ginecológicas, etc.).
Farmacocinética.
Como todos los betalactámicos poseen una excelente difusión a órganos (vesícula biliar, próstata, piel, pulmón, riñón) y líquidos orgánicos (esputo, orina, secreción bronquial, líquido peritoneal, sinovial, pericárdico, ascítico, semen). Con meninges inflamadas alcanza concentraciones terapéuticas en el SNC.
La biodisponibilidad por vía oral de aztreonam es menor del 1 %; es utilizado como preparado para uso intravenoso o intramuscular y alcanza altas concentraciones plasmáticas en 30 minutos. Su vida media es 1,3 a 2 horas; en la ancianidad es de 2,7 horas aproximadamente, así como en enfermedades hepáticas; penetra bien en todos los tejidos incluso en las meninges, aun en ausencia de inflamación; también en tejido sinovial, hueso, próstata, en tracto respiratorio y líquidos corporales. Se excreta por el riñón.
Algunos parámetros farmacocinéticas de Monobactámico

Efectos indeseables.
Hipersensibilidad: anafilaxia, andioedema, broncoespasmo.
Piel: exantema, prurito, urticaria, eritema multiforme, y dermatitis exfoliativa.
Hematológicos: eosinofilia, trombicitosis, trombocitopenia, leucosis, neutropenia, anemia, pancitopenia.
Hepatobiliar: elevaciones ocasionales de las transaminasas hepáticas y de la fosfatasa alcalina.
TGI: náuseas, vómitos, diarreas, calambres abdominales, úlceras orales y alteración del gusto. Provoca enterocolitis pseudomembranosa.
Debe ser utilizado con precaución en la insuficiencia renal, aun cuando no es primariamente nefrotóxico.
Interacciones medicamentosas.
La admistración concomitante con probenecid o furosemida causa un incremento significativo en el suero de aztreonam.
Usos de los monobactámicos:
Son útiles en las sepsis por gram (-) como:
Infecciones respiratorias bajas nosocomiales o en pacientes inmunocomprometidos (EPOC, fibrosis quistica, diabéticos.
Sepsis urinaria complicadas y no complicadas.
Sepsis ortopédicas. Hay que tener en cuenta la frecuencia de la flora gramnegativa que invade el hueso en la sepsis posprótesis y en las fracturas de origen traumático, así como en la artritis piógena.
Sepsis del sistema nervioso central. Por gérmenes gramnegativos solo o combinado con otros antibacterianos. Septicemia. Ha sido eficaz para el tratamiento por E. Coli, K. pneumonia, Enterobacter, P. aeroginosa, P. mirabilis, S. marcescens, Citrobacter o h. influenzae susceptibles.
Infecciones intraabdominales y del aparato reproductor femenino. Junto con otros antimicrobianos puede utlizarse para tratar infecciones intraabdominales por Enterobacteria y P. aeroginosa. En forma semejante, puede emplearse para tratar infecciones en las vías genitales de la mujer, incluidas la endometritis o inflamación pélvica.
Gonorrea. Basta una dosis de 1 g de aztreonam, para tratar blenorragia uretral, endocervical, anorrectal o de los tres tipos, no complicadas, causadas por N. Gonorrhoeae, produzca o no penicilinasa.
Otras infecciones. Infecciones biliares, quemaduras, tratamiento de las úlceras de decúbito u otras infecciones por gérmenes gram (-) que infectan la piel, aunque debe asociarse a otros antimicrobianos cuando se sospecha la presencia de gram (+) y anaerobios.

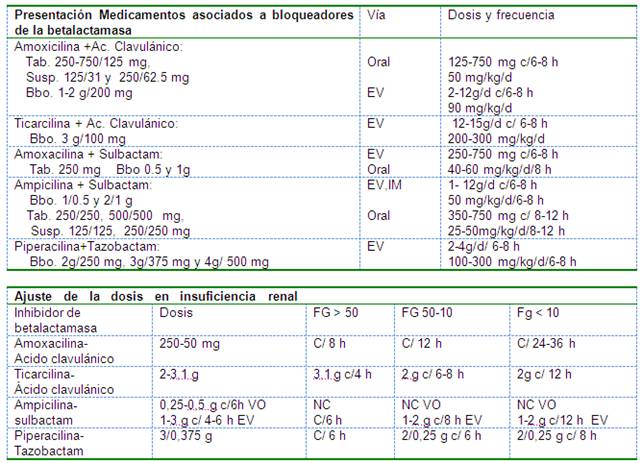
E. Asociados a Inhibidores de (-lactamasa
Historia.
Este grupo surgió como una estrategia para enfrentar la resistencia de los microrganismos a los betalactámicos ocasionada por las betalactamasas, enzimas que hidrolizan el enlace amida del anillo betalactámico y lo inactivan.
Antes que la penicilina G fuera utilizada clínicamente, EP. Abraham y E. Chain, habían descrito en 1940, la existencia de enzimas betalactamasas. Cuatro años más tarde, Kirby estableció la presencia de estas enzimas como mecanismo de resistencia del Staphylococcus aureus, que en esa época comenzaba a aparecer, pero que ya a la altura de 1952, el 75 % de las cepas intrahospitalarias poseían resistencia por esta vía.
Durante años los investigadores se dieron a la tarea de buscar sustancias que fueran capaces de inhibir estas enzimas inactivadoras de antibióticos. Una serie de compuestos provenientes de cepas de Streptomyces olivaceus, a los cuales denominaron ácidos olivánicos, fueron encontrados con dichas propiedades, sin embargo su rápida metabolización no permitió su amplia utilización.
En 1973 es detectado otro grupo de sustancias con potente acción inhibitoria sobre las betalactamasas; estas se obtuvieron a partir de cepas de Streptomyces clavuligerus y se denominaron clavamas; las mismas tienen una estructura betalactámica con escasa actividad antibacteriana pero un amplio espectro antienzimático. Tres o cuatro años demoraron en determinar su estructura bioquímica y sus propiedades microbiológicas; en 1976 Browm y col. informaban sobre dicho hallazgo, apareciendo el primero y más potente inhibidor de betalactamasas, el ácido clavulánico. Posteriormente aparecieron otros compuestos semisintéticos derivados del ácido penicilánico, con excelente actividad inhibitoria de betalactamasas; sulbactama y tazobactama.
Clasificación
1. Clavamas: Ácido clavulánico.
2. Derivados del ácido penicilánico: Sulbactam, Tazobactam, Brobactam.
3. Carbapenemas: Ácido olivánico, Tienamicinas, Epitienamicinas, Asparenomicinas, Carpetimicinas.
4. Derivados del ácido heptonoico: Cilastatina
5. Combinadas con diferentes betalactámicos:
Amoxacilina + Ácido clavulánico (Augmentin).
Ticarcilina + Ácido clavulánico (Timetin).
Amoxacilina + Sulbactam (Trifamox)
Ampicilina + Sulbactam (Unasyn).
Piperacilina + Tazobactam (Zosin)
Estructura química
El ácido clavulánico es un betalactámico, específicamente un oxapenema, donde el anillo secundario en vez de ser tíazolidina es de oxazolina y carece de cadenas lateral en posición 6. El sulbactam y el tazobactam son sulfotas del ácido penicilánico, también de estructura betaláctamica.
Mecanismo de acción.
Los inhibidores de betalactamasas actúan sobre los betalçactamicos mediante un mecanismo de inhibición que se caracteriza por ser competitivo, progresivo e ireversible. Teniendo una estructura semejante a los betalactámicos, compiten con los mismos por unirse al sitis de activo de la betalactamasa, siendo reconocidos por estas últimas como substrato prioritario dando lugar a una unión inactivadota. Algunos inhibidores se fijan a la enzima un tiempo suficiente para que el antimicrobiano actúe sobre la bacteria y un grupo de ellos, llamados inactivadotes suicidas (ácido clavulánico), se fijan de forma irreversible.
Espectro de acción
El ácido clavulánico, el sulbactam y el tazobactam tienen poca actividad antibiótica propia, pero pueden ampliar el espectro del antimicrobiano con el cual se asocia permitiendo controlar con monoterapia y a dosis inferiores muchas infecciones. En combinación con ampicilina, amoxicilina, ticarcilina o piperacilina, esos fármacos aumentan la efectividad contra ciertos gérmenes que son resistentes, además el sulbactam tiene un efecto bactericida superior al tazobactam y cefalosporinas frente al Acinetobacter sp., microorganismo implicado en brotes de infecciones nosocomiales en las unidades de cuidados intensivos.
La adición de ácido clavulánico o sulbactam a la ampicilina o la amoxicilina, aumenta la actividad contra estafilococos productores de betalactamasa, Haemophilus influenzae, Neisseria gonorrhoeae, Moraxella catarrhalis, Klebsiella sp., Bacteroides y otros anerobios.
La asociación ticarcilina más ácido clavulánico tiene un espectro de actividad más amplio que la ticarcilina sola y resulta eficaz contra Neisseria gonorrhoeae, estafilococos y Haemophilus influenzae, así como contra especies de klebsiella, serratia y bacteroides. La
piperacilina más tazobactam tiene un espectro superior con respecto a la piperacilina, incluye a Neisseria gonorrhoeae, estafilococos, Haemophilus influenzae y especies de bacteroides, así como una mayor actividad frente a klebsiella y serratia. Estas asociaciones no son más eficaces contra Pseudomona aeruginosa que la ticarcilina o la piperacilina sola.
Mecanismos de resistencia
Los inhibidores de betalactamasas tienen acción sobre la mayoría de las betalactamasas codificadas por plásmidos, careciendo de efectividad sobre las betalactamasas cromosómicas inducibles, de la clase Ia perteneciente a la clasificación de Richmond y Sykes, que son predominantemente cefalosporinasas elaboradas por bacilos gramnegativos aerobios (Morganella morganii, Providencia rettgeri, Enterobacter sp).
En la década del 90 fueron reportadas un grupo de betalactamasas resistentes al ácido clavulánico y al sulbactam. Hasta el momento se han descrito un total de 17 tipos, presentes principalmente en E. coli y K. pneumoniae, permaneciendo sensibles solo al tazobactam.
Otras enzimas resistentes a estos inhibidores de betalactamasas, surgidas por el uso repetitivo de las oxiiminocefalosporinas y presentes en Enterobacter, Citrobacter, Pseudomona, E. coli y K. pneumoniae son las llamadas AmpC, de las cuales sólo el tipo CMY son inhibidas por tazobactam.
Farmacocinética
La farmacocinética de los inhibidores de betalactamasas deben corresponderse con la de los antimicrobianos betalactámicos con los que se asocian (ampicilina, amoxicilina, ticarcilina, piperacilina), presentando una buena difusión a órganos, tejidos y líquidos orgánicos, aunque el clavulámico no alcanza concentraciones efectivas en el SNC. El clavulámico y el sulbactam se absorben por vía oral, el tazobactam se administra solo por vía parenteral. Se eliminan principalmente por vía renal. La excreción urinaria del ácido clavulánico es del 40-50 %, a través del filtrado glomerular, mientras que la del sulbactam es de 80-85 % y el tazobactam del 60-65 %, ambos tanto por filtrado como por secreción tubular.
Efectos adversos
Son comunes a los de los betalactámicos; las asociaciones con las penicilinas pueden provocar todas las reacciones mencionadas para ellas en específico.
Se han descrito alteraciones cutáneas como el rash urticariano propias de la hipersensibilidad causada por los antimicrobianos betalactámicos, por lo que debe explorarse el antecedente de reacciones alérgicas previas. También se han reportado trastornos gastrointestinales fundamentalmente diarreas.
No deben ser utilizados durante el embarazo y la lactancia.
Caracteristicas generales de los principales medicamentos
Ácido clavulánico. Se absorve adecuadamente depués de ingerido y también puede aplicarse por vía parenteral (EV). Se le ha combinado con la amoxacilina en un preparado oral (Augmentin) y con la ticarcilina en un preparado parenteral (Timetin). La combinación de Amoxacilina + ácido clavulánico extiende su espectro al grado que se asemeja al imipenem y en su acción incluye bacilos gramnegativos aerobios (H. influenzae, N. Gonohrroreae y E. Coli), Staphylococcus aureus y especie de bacteriodes. No se advierte mayor acción contra especie de Pseudomona. La combinación es útil en infecciones nosocomiales mixtas y a menudo se utiliza junto con un aminoglucósido.
Sulbactam. Es semejante en estructura al ácido clavulánico. Puede ingerirse o aplicarse por vía parenteral junto con un antimicrobiano (-lactámico. Se le distribuye para su uso intravenoso o intramuscular en combinación con ampìcilina (Unasyn). Esta combinación posee actividad satisfactoria contra cocos grampositivos que incluyen las cepas de Staphylococcus aureus productora de (-lactamasa, aerobios gram (-), (pero no pseudomona) y anaerobios; tambiém se ha utilizado para tratar infecciones intraabdominales y pélvicas mixtas.
Tazobactam. Es un inhibidor de las sulfonas de (-lactamasa del ácido penicilánico. En común con otros inhibidores disponibles, de poca acción contra (-lactamasa cromosómicas inducibles de Enterobacteriaceae, pero tiene actividad satisfactoria contra muchas de las (-lactamasa de plásmido que incluyen algunas de las de espectro extendido. Se ha combinado con Piperacilina en la forma de un preparado parenteral (Zosyn). La combinación no aumenta el efecto frente a Pseudomona aeruginosa y puede ser inefectiva en algunas ocasiones, debido a que la dosis recomendada de la combinación es menor que la piperacilina sola. La combinación de Piperacilina + tazobactam tiene espectro similar a la combinación Ticarcilina + ácido clavulánico.
Aplicaciones clínicas
Debido a los crecientes niveles de resistencia bacteriana en el momento actual y por ser el mecanismo principal la producción por las bacterias de enzimas inactivadoras de betalactámicos (betalactamasas), son estas combinaciones, de inhibidores con betalactámicos, los medicamentos ideales para contrarrestar este fenómeno. Combinaciones como amoxicilina/ácido clavulánico, piperacilina/tazobactam, ticarcilina/ácido clavulánico, y tienamicina/cilastatinas se encuentran en la era avanzada de la terapia para enfrentar la sepsis del paciente crítico.
Debido a la gran participación de gérmenes productores de betalactamasas: Haemophilus influenzae, Moraxella catarrhalis, Staphylococcus aureus, Neisseria gonorrhoeae, Bacteroides fragilis, etc., son de gran utilidad en el tratamiento de: sinusitis, otitis medía, mastoiditis, epiglotitis, uretritis, neumonías nosocomiales, bronquitis crónicas infectadas, osteomielitis, infecciones hepatobiliares, sepsis abdominal, peritonitis, infecciones cutáneas, ginecológicas y en la profilaxis quirúrgica. Por su escasa difusión al sistema nervioso central, no tiene indicación en la sepsis a este nivel.
En estas combinaciones la dosis se calcula de acuerdo con el antimicrobiano base es decir la ampicilina, la amoxicilina, la ticarcilina o a la piperacilina.
Cetólidos
Los Cetólidos son un nuevo grupo de antibioticos que están estructuralmente relacionados a los macrólidos. Ejemplos: Telitromicina (Ketek) y Cetromicina
Fuente y Química.
La telitromicina (KETEK) es el único cetólido actualmente aceptado, es derivado semisintético de la eritromicina, difiere de ella, en que un grupo 3-ceto reemplaza la cladinona cerca del macrolido de 14 átomos (eritromicina A), y hay un carbamato sustituido en C11-C12. Estas modificaciones dan al cetólido menos susceptible a metilarse, siendo más resistente, y tiene un espectro más amplio de actividad. Fue aprobada por FDA en el 2003. Ver la fórmula estructural de la telitromicina más adelante.

Actividad antibacteriana.
Cetólidos y macrólidos tienen las propiedades antibacterianas muy similares. Telitromicina es activo contra: S. neumococos (incluyendo penicilina resistente, macrólidos resistentes y multirresistentes), H. Influenzae, M. Catarrhalis, clamidias, legionellas, mycoplasmas y anaerobios, exceptuando B. fragilis.
El mecanismo de Acción y Resistencia.
Cetólidos y macrólidos tienen el mismo el sitio designado, actúan inhibiendo la síntesis proteica a nivel ribosomal, específicamente, bloqueando la elongación proteica por inhibición de la peptidil-transferasa. La diferencia principal entre los dos es las modificaciones estructurales dentro del cetólidos neutraliza los mecanismos de resistencia comúnes que hacen a los macrólides ineficaz. La introducción de los 3-ceto convertido de la función un macrolido metilasa-induciendo en un cetólido no inducido. Esta mitad también previene la salida de la droga, probablemente porque genera un substrato menos deseable. La substitución del carbamate a C11-C12 refuerza ligando al ribosomal al sitio designado, incluso cuando el sitio es metilado, introduciendo una interacción extra del cetólido con el ribosoma. Las tensiones inducidas por los productos de la metilación constitutivas de S. los pneumoniae son por consiguiente Inducible y susceptibles a la telitromicina; y sin embargo, las tensiones constitutivas producto de metilación de S. el aureus y S. los pyogenes son telitromicina-resistentes porque la fuerza de la interacción del cetólido con el ribosomal es sitio obligatorio de la metilación el es la resistencia superada insuficiente.
Farmacocinetica
La Telitromicina se formula como una forma rápida para la administración oral de 400-mg No hay ninguna forma parenteral. Es bien absorbida con aproximadamente 60% biodisponibilidad. Las concentraciones de suero máximas, promediando 2 mg/ml que siguen tras una sola dosis oral de 800-mg, se logra dentro de 30 minutos a 4 horas. Con una vida media de 9.8 horas, la droga puede darse una vez al día. El 60% a 70% es ligada por las proteínas del suero, principalmente la albúmina. Penetra bien en la mayoría de los tejidos, mientras excediendo las concentraciones del plasma por aproximadamente dos – a cuatro o más. Telitromicina se concentra en los macrófago y las células de sangres blancas dónde las concentraciones de 40 mg/ml (500 veces la concentración del plasma simultánea) se mantiene 24 horas después de dosificar. La droga se aclara principalmente por el metabolismo hepático, 50% por CYP3A4 y 50% por el metabolismo CYP-independiente. No necesita ajuste de dosis para la insuficiencia renal o hepática ligera a moderada. Ninguna dosis se ha establecido para pacientes en quienes el aclaramiento de la creatinina está menos de 30 ml/minuto, aunque una reducción en la dosificación probablemente es aconsejable.
Efectos indeseables.
Generalmente se tolera bien.
Las náuseas, vómitos, y las diarreas son lo que más común efectúa, mientras ocurriendo en 3% a 10% de cursos del tratamiento.
Las perturbaciones visuales debido al alojamiento retardado ocurren en aproximadamente 1% de cursos del tratamiento, e incluya visión borrosa, dificultad enfocando, y diplopia.
Trastorno hepático reversible con elevación de las transaminas, se han informado hepatitis y hepatotoxicidad severo (INSUFICIENCIA HEPATICA)
La colitis Pseudomembranoass se ha informado.
No se recomienda para el uso de la rutina en los pacientes con miastenia gravis, debido a los informes de exacerbación de la enfermedad en los pacientes tratados.
Puede causar la prolongación de QTc significativo y riesgo aumentado de arritmia ventricular clínicamente en los pacientes predispuestos.
No debe usarse en los pacientes con el síndrome de QT prolongado, hipokalemia o hipomagnesemia, bradicardia profunda, o en pacientes que reciben cierto antiarritimicos (por ejemplo, quinidine, procainamide, amiodarone) u otros agentes que prolongen el QTc (e.g., cisapride, pimozide).
Interacciones
Telitromicina tiene algunos interacciones clínicamente significantes con drogas similar a la eritromicina. Es un substrato y " un inhibidor fuerte de CYP3A4. La coadministración con rifampina, un inducyor potente de CYP, disminuye las concentraciones de suero de telitromicina un 80%. Los inhibidores de CYP3A4 (por ejemplo, itraconazole) incrementan concentración sérica de teliromicina. Las concentraciones de suero de substratos de CYP3A4 (por ejemplo, pimozide, cisapride, midazolam, estatinas, cyclosporine, fenitoina) incrementan por la telitromicina. La Telitromicina también aumenta concentraciones en suero máximas de metoprolol y digoxina.
Usos terapéuticos.
Dado su espectro de actividad es aceptado para el tratamiento de infecciones del tracto respiratorias, incluyendo exacerbación agudo de bronquitis crónica (régimen de 5 días), sinusitis bacteriana aguda (régimen de 5 días), y la neumonía adquirida en la comunidad (régimen de 7 a 10-días).
Aunque la telitromicina no se indica para el tratamiento de neumonía severa o bacteriemia, se curaron casi el 90% de pacientes que demostrados tener clínicamente bacteriemia por pneumococco después de tomarlo.
En los ensayos premarketing de la telitromicin en los pacientes con neumonía adquirida en la comunidad causados por múltiple-droga-resistentes de S. pneumoniae (resistente a las penicilinas, cefalosporinas, macrolidos, tetraciclinas, o trimethoprim-sulfametoxazole) por encima del 90% de los pacientes se curó.
Por su excelente penetración intracelular son efectivos en la sepsis causadas por gérmenes intracelulares (clamidias, legionelas, micoplasmas) y tienen buen efecto frente a H. influenzae.
Dosis recomendada:
Telitromicina 800 mg monodosis diaria, de 5 – 10 días (oral).
Diaminopiridinas
Son antimicrobianos bacteriostáticos que actúan alterando el metabolismo del ácido fólico en la bacteria. Su espectro de acción es similar al de las sulfonamidas. Inicialmente se usó a dosis tóxicas, pero después se observó que, asociada a una sulfamida, producía efectos sinérgicos. Desde entonces se emplea preferentemente en combinación fija con el sulfametoxazol (cotrimoxazol) y, en algunos países, con sulfadiazina (cotrimazina) y sulfamoxol (cotrifanol). Ver asociaciones de las sulfas.
Trimetoprima (TMP): Es una 2,4-diaminopirimidina.

Mecanismo de acción
Inhibe la dihidrofolato-reductasa de bacterias y protozoos, con una sensibilidad 50.000-100.000 veces superior que la enzima de células humanas. De este modo interfiere en la transformación de dihidrofolato en tetrahidrofolato y, secundariamente, en la síntesis de ácido desoxitimidílico, resultando en una inhibición de la síntesis de ADN y proteínas bacterianas.
Actividad antibacteriana
Es un fármaco bacteriostático. In vitro es activo frente a la mayoría de bacterias: cocos grampositivos (Staphy- lococcus, S. pyogenes, Streptococcus viridans, S. pneu- moniae, Corynebacterium diphtheriae) y bacilos gramne- gativos, exceptuando P. aeruginosa y Bacteroides sp. La mayoría de los anaerobios, Treponema pallidum, M. tu- berculosis y Mycoplasma sp son resistentes.
Mecanismos resistencias bacterianas.
Las resistencias bacterianas a trimetoprima se deben a cambios en la permeabilidad celular, a una disminución de la capacidad de fijación fármaco-bacteria o a una superproducción o alteración de la enzima dihidrofalato-reductasa. La alteración enzimática está codificada por un plásmido (factor R), mientras que los restantes mecanismos se deben a mutaciones cromosómicas. El mecanismo más importante por su repercusión clínica es el debido a la existencia del plásmido.
Características farmacocinéticas
Se absorbe rápidamente de forma casi completa por vía oral (85-90 %). La Cmáx se alcanza 1-4 horas después de su administración; es aproximadamente de 1 &µg/ml a la dosis de 100 mg y entre 2-4 &µg/ml con 160 mg. Puede administrarse en infusión IV, alcanzándose la Cmáx 1 hora después con una concentración de 3,4 &µg/ml a la dosis de 160 mg. La distribución tisular es amplia, alcanzando niveles superiores a los sanguíneos en riñón, orina, pulmón, esputo, saliva, leche, hígado, bilis, próstata, secreción prostática y vaginal. Atraviesa la barrera placentaria. En el LCR alcanza el 40-50 % de los niveles sanguíneos. La semivida de eliminación es de 9-11 horas aumentando en caso de insuficiencia renal. Estudios farmacocinéticos en niños sugieren la variación de la semivida dependiendo de la edad.
La trimetoprima es metabolizada en el hígado aproximadamente en el 20 % de la dosis administrada, originando cinco metabolitos: oxi, hidroxi, carbonilo y dos desmetilados. Todos, excepto el derivado hidroxi, mantienen actividad bacteriostática. Se excretan por orina y bilis. El 60-80 % de la dosis de trimetoprima se elimina en orina de 24 horas mediante filtración glomerular y secreción tubular. El componente de reabsorción depende del pH urinario, siendo bloqueado en condiciones de acidez. La concentración alcanzada en orina es 100 veces superior a la sérica; la mayor parte es trimetoprima original, el 8 % en forma conjugada. Una pequeña proporción se excreta por bilis, en cantidad suficiente para reducir o eliminar la flora fecal susceptible que interviene con frecuencia en el origen de las infecciones.
Reacciones adversas e interacciones
Es un fármaco que produce pocas reacciones adversas. Se han descrito reacciones de hipersensibilidad (dermatitis exfoliativa, eritema multiforme, síndrome de Stevens-Johnson, síndrome de Lyell y anafilaxia) e interferencia en la hemopoyesis (trombocitopenia, leucopenia, neutropenia, anemia megaloblástica y metahemoglobinemia) en pocas ocasiones, sobre todo en tratamientos prolongados y dosis altas.
Se han observado también otras alteraciones dermatológicas, como prurito, fototoxicidad y erupciones de tipo maculopapular morbiliforme y pruriginoso que aparecen generalmente entre el 7° y el 14° día de tratamiento y cuya frecuencia es del 3-7 %. En cuanto al tracto gastrointestinal, pueden observarse molestias gástricas, náuseas, vómitos y glositis. Puede producir aumento de transaminasas y bilirrubina; con poca frecuencia, ictericia colestásica.
La trimetoprima puede inhibir el metabolismo hepático de la fenitoína, aumentando su semivida en el 51 %. Se ha observado sinergia con polimixina, rifampicina y metronidazol.
Aplicaciones terapéuticas
Está indicada en el tratamiento de infecciones urinarias no complicadas producidas por bacterias sensibles, a dosis de 100 mg cada 12 horas o 200 mg una vez al día durante 10 días. Está contraindicada en personas con hipersensibilidad al fármaco y en caso de anemia megaloblástica debida a déficit de folato. Es recomendable evitarla durante el embarazo y en los primeros meses de vida. Su administración debe ser controlada en caso de in- suficiencia renal. Si el aclaramiento de creatinina es de 15-50 ml/min, la dosis debe reducirse a 50 mg cada 12 horas.

Pirimetamina (Daraprim) (Ver antipalúdicos)
Presentación y dosis
Tableta 25 mg.
Posología:
Toxoplasmosis:
Adultos: pirimetamina (50 a 200 mg/d) con sulfadiacina (250 a 100 mg c/6 h) por 1 ó 2 d seguido de pirimetamina 25 a 50 mg/d con sulfadiacina 125 a 500 mg c/6h por 2 a 4 semanas en pacientes inmunocompetentes, o de 4 a 6 en inmunocomprometidos.
Niños: 1mg/kg/d por 1 a 3 d, seguido de 0.5 mg/kg/d por 4 a 6 semanas, debe administrarse las dosis usuales de sulfadoxina.
Isosporiasis: Tratamiento de mantenimiento en pacientes con VIH: 50 a 75 mg.
Asociadas.
Tableta Pirimetamina 25 mg / Sulfadoxina 500 mg (FANSIDAR)
Posología:
Isosporiasis: Tratamiento de mantenimiento en pacientes con VIH: Pirimetamina 25 mg con sulfadoxina 500 mg semanalmente.
Estreptograminas
Historia.
En 1953, a partir de una muestra de suelo, fue aislado un nuevo Streptomyces
(graminofaciens), el cual era capaz de producir un grupo de sustancias
antibióticas, que fueron nombradas estreptograminas: Micamicina, pristinamicina,
vernamicina y virginamicina. En los últimos años se desarrolló
una investigación sobre un derivado semisintético de esta familia,
una asociación de quinupristina-dalfopristina, el cual fue aprobado por
la FDA de los EE.UU en 1999.
Fuente.
Las pristinamicinas pertenecen al grupo terapéutico de las estreptograminas.
De la pristinamicina IA deriva la quinupristina, que es una macrolactona péptido
(estreptogramina A). Mientras que la dalfopristina deriva de la pristinamicina
IIB y es una lactona poliinsaturada (estreptogramina B). Ver estructuras químicas
más adelante.

Mecanismo de acción.
Las estreptograminas inhiben la síntesis de proteínas al unirse
a la subunidad ribosomal 50S. Un derivado, la quinupristina, inhibe la elongación
de la cadena polipéctidica y la dalfopristina interfiere directamente
con la peptidil transferasa.
Espectro.
Bacterias gram (+) resistentes a multiples drogas: estafilococos aureus
resistentes a meticilina, estafilococos coagulasa negativos, enterococo
vancomicina resistente, E Faecium sensibles y resitentes a vancomicina
y teicoplanin, S viridans, S pyogenes, Lactobacillus,
Bacteroides, Prevotella, Listeria monocytógenes,
Legionella, Neumococo penicilina resistente, eritromicina resitente y multirresistente,
Mycoplasma penumoniae, moraxella catarrhalis. También tienen actividad
in vitro frente a gonococo, meningococo, Chamydia trachomatis,
Mycoplasma hominis.
Resistencia antimicrobiana: Se han descrito 3 mecanismos de resistencia:
1-Disminución de la afinidad de la quinupristina por el locus ribosómico,
al cual se une para realizar su acción. 2- A través de una enzima
acetiltransferasa y 3- Por el bombeo activo bacteriano. Algunos estudios han
culpado al elevado uso de la virginimicina en la alimentación avícola
como responsable de la reciente resistencia a este compuesto.
Farmacocinética. A diferencia de las pristinamicinas, los derivados
quinupristina y dalfopristina pueden administrarse por vía EV, ya que
son solubles en agua. Poseen actividad antibacteriana limitada que aumenta notablemente
cuando se emplean de manera sinérgica y se combinan en la proporción
30:70 (Q-D). Poseen amplia distribución tisular y unión a proteínas
plasmáticas. Rápida eliminación. No atraviesan la barrera
hematoencefálica ni la placentaria en grado significativo. Presentan
excreción biliar primaria. Son convertidas en metabolitos activos a nivel
hepático. Se excretan por vía renal el 15-19%. Son captados por
los macrófagos y activos intracelularmente. El tiempo de vida media es
de 1 hora (Q) y de 0,75 horas (D).
Efectos adversos.
En ensayos clínicos los más frecuentes fueron: dolor, inflamación
y flebitis en el sitio de inyección, náuseas, vómitos,
diarreas, artralgias, mialgias, rash. Otros: aumento de la creatinina sérica,
trombocitopenia, anemia, eosinifilia, aumento de la bilirrubina conjugada.
Indicaciones:
Sobre todo en infecciones por estafilococo aureus resistentes a meticilina
y enterococos faecium sensibles y resitentes a vancomicina y teicoplanina

Fenicoles
Historia.
El cloranfenicol o cloromicetina fue aislado a partir del actinomiceto Streptomyces
venezuelae en 1947 por Ehrlich y Burkholder. Al principio este compuesto
despertó un gran interés, debido a que poseía en apariencia
una toxicidad relativamente escasa, un amplio espectro de actividad antimicrobiano
y una gran efectividad terapéutica al administrarlo por la vía
oral en una serie de enfermedades.
El entusiasmo por su amplia actividad antibacteriana fue decayendo a partir
de que se publicó el primer caso de anemia aplástica por cloranfenicol
en 1950 por Rich y colaboradores, y de las diversas discrasias sanguíneas
graves descritas en esa mima década.
En 1952 Cutler y col, reemplazaron el grupo nitro por un grupo sulfometil y
sintetizaron un derivado del cloranfenicol, el tianfenicol. Con la aparición
de cepas Haemophilus influenzae resistentes a las peniclinas y al mejor
conocimiento de los gérmenes anaeróbicos como el Bacteroides
fragilis, se ha aumentado notablemente la frecuencia de su utilización
en la actualidad.
Química.
El cloranfenicol es un compuesto liposoluble derivado del ácido dicloroacético, constituido por un anillo aromático (nitrobenceno) y una cadena alifática (Ver estructura química).

En el empeño de encontrar nuevos congéneres del cloranfenicol con mayor o igual actividad y menor toxicidad se han ensayado numerosos compuestos, pero los resultados no han sido favorables. Sólo se describe al tianfenicol, el cual presenta un radical sulfometilado en lugar del grupo nitro del benceno, pero su actividad antimicrobiana es menor que la del cloranfenicol.

Espectro antimicrobiano.
Es de amplio espectro frente a un gran número de bacterias gram (+) y negativas, tanto aeróbicas como anaeróbicas, micoplasma, ricketsias, clamidias y treponemas. Gram (+): Streptococo (pneumoniae, pyogenes, agalactiae), enterococos. Gram (-) (N. meningitidis, H. Influenzae, Salmonella typhi, Shigella, Vibrio cholera, Brucella). Anaerobias: (Bacteroides fragilis, Clostridium). Su actividad puede ser bactericida (N. Meningitidis y H. Influenzae) y para el resto bacteriostática.
Mecanismo de acción:
Interfiere en la síntesis proteica bacteriana al fijarse a la subunidad ribosomal 50S, en el sitio de la peptidil transferasa y bloquea la reacción de traspetidación, por lo que no ocurre la formación de enlaces peptídicos. Este mecanismo de acción explica el antagonismo que se produce al asociar cloranfenicol con los macrólidos (eritromicina), lincomicidas (lincomicina y clindamicina), pues todos estos antimicrobianos ejercen sus efectos al unirse a la misma fracción ribosomal 50 S. También es capaz de inhibir la síntesis de proteínas en las células de mamíferos, principalmente en los ribosomas mitocondriales (por su semejanza estructural). Particularmente sensible a esta acción son las células eritropoyéticas, lo que explica en parte sus principales toxicidades.
Resistencia antimicrobiana:
Esta dada por pérdida de la permeabilidad de la membrana bacteriana y a la producción bacteriana de una enzima acetiltranferasa, la cual causa acetilación de los fenicoles convirtiendolos en productos inactivos.
Farmacocinética.
Buena absorción oral, puede ocurrir absorción sistémica después de su uso oftálmico. Activo por vía oral e intravenosa. Buena distribución en los fluidos corporales incluído LCR. Atraviesa la barrera placentaria y se encuentra en la leche materna. Buena distribución en los tejidos. Metabolismo Hepático. Excreción renal inactivo, pequeñas proporciones se excretan en forma activa y sirven para el tratamiento de infecciones del TU. Vida Media: 1,6-4,1hs. En enfermedades hepáticas se prolonga hasta 3-12hs. Debido a la deficiencia en la glucoroconjugación, baja hidrolísis hepática y disminución de la eliminación renal, el Cloranfenicol debe usarse monitoreado en los neonatos.
Algunos parámetros farmacocinéticas de los Fenicoles

Reacciones adversas.
1. Discrasias sanguíneas:
a) Depresión reversible de la médula ósea (Dosis dependientes)
b) Aplasia medular (idiosincarasia) generalmente irreversible, puede llevar a la muerte hasta el 80% de los casos. Se presenta con una incidencia de 1:50 000 y 1:100 000 casos tratados y no guarda relación con la dosis. c) Anemia hemolítica en personas con défícit de glucosa 6 fofato deshidrogenasa (G6PD) del tipo mediterráneo.
2. TGI: Glositis, náuseas, vómitos, diarreas (Suprainfecciones).
3. Neurológicas: Neuritis óptica, Cefalea, Delirium.
4. Hipersensibilidad: fiebre, exantema, angioedema, urticaria y anfilaxia.
5. Síndrome del Bebé Gris: en prematuros y recién nacidos (por la falta de desintoxicación metabólica por la inmadurez enzimática y una dosificación elevada).
Interacciones medicamentosas.
Cloranfenicol. Aumenta los niveles de: Hipoglicemiantes orales, Fenitoína, Ciclofosfamida, Warfarina. Aumentan el metabolismo del Cloranfenicol: Fenobarbital, Rifampicina, Fenitoína.
Contraindicaciones.
En individuos con antecedentes de hipersensibilidad o reacciones tóxicas al cloranfenicol. No utilizar en el tratamiento de infecciones triviales ni como agente profiláctico para evitar infecciones bacterianas en: embarazo y lactancia, en recién nacidos y prematuros ni en discrasias sanguíneas.
Indicaciones:
Es importante utilizar sólo el Cloranfenicol en infecciones en que sus beneficios excedan los riesgos de toxicidades posibles. Si se dispone de otros antimicrobianos igualmente eficaces y que puedan ser menos tóxicos que el cloranfenicol, deben utilizarse preferiblemente: Fiebre tifoidea o paratifidea (infecciones graves por Salmonella.)
Meningitis: H. Influenzae resistente a ampicillina o cuando no se puede utilizar ésta o una cefalosporina de tercera generación y en meningitis por neumococo o meningococo en pacientes alérgicos a la penicilina. Infecciones anaerobias.
Sepsis grave por Bacteroides fragilis, incluyendo el SNC (Absceso encefálico en combinación con la penicilina).
 Página anterior Página anterior | Volver al principio del trabajo | Página siguiente  |