Conglomerados de Sulfuro en la Síntesis del Hem (Bioquímica) (página 2)
El presente trabajo recoge
información reciente de diversas
publicaciones y trabajos realizados con el propósito de
brindar información muy detallada para aquellas personas
interesadas en conocer la causa de la anemia
sideroblastica. Además este trabajo refleja una base
biográfica de esta patología.
Objetivos
1. OBJETIVO GENERAL
Analizar y comprender la nueva via de los Centros o
conglomerados de Sulfuro de Hierro para la síntesis
del grupo HEM de la hemoglobina.
2. OBJETIVOS ESPECIFICOS
Conocer la importancia del hierro para la síntesis
del HEM.Explicar las reacciones de la síntesis del grupo
HEM de la hemoglobinaConocer los mecanismos de regulación para la
síntesis del HEMAnalizar y explicar la composición, estructura y
función del grupo HEM de la hemoglobinaExplicar los mecanismos por lo que los Centros de Sulfuro
de Hierro y las proteínas Reguladoras de Hierro Tipo
1, intervienen en la regulación de la síntesis
del HEMExplicar los mecanismos patógenos que origina la
deficiencia o alteración en la síntesis del
HEM
Marco
referencial
El hierro es el
cuarto elemento más abundante en la tierra, su
absorción ocurre en el duodeno y yeyuno superior, el
ácido clorhídrico favorece la reducción de
este catión a la forma ferrosa. El transportador de
metales
divalentes-1 DMT1, interviene en el transporte de
hierro desde la luz intestinal al
entericito, una vez que el hierro entra a la célula,
la ceruloplasmina se encarga de oxidar el Fe+2 a Fe+3, para que
así sea captado por la apotransferrina, que se transforma
a transferrina. Su transporte es realizado por la transferrina
(TF).
La captación celular del hierro se efectúa
mediante un receptor de transferrina (RTf). el hierro excedente
se almacena en el citoplasma unido a la ferritina.
Proteínas Reguladoras Del Hierro
Las proteínas
reguladoras de hierro (conocidas por sus siglas en inglés,
IRP), son proteinas citoplasmáticas capaces de unirse a
los elementos de respuesta al hierro o IRE (del inglés
iron responsive elements) de los ARN mensajeros (ARNm)
de las proteínas implicadas en el metabolismo
del hierro, para así controlar la síntesis
del receptor de transferrina, DMT1 y de ferritina.
Esta proteína posee un conglomerado o centro del
sulfuro de hierro (4Fe-4S) que le permite cambiar entre dos
actividades diferentes en dependencia del nivel de hierro
celular.

Así, cuando la concentración del hierro es baja,
IRP-1 no es limitado por el hierro y es capaz de atar a extremo
5' del RNA del mensajero de la ferritina en un sitio llamado el
elemento regulador del hierro, un proceso que
bloquea la traducción del mRNA. Inversamente, cuando
la concentración del hierro es alta, y la célula
necesita ferritina, el IRP-1 ata el hierro y llega a ser
incompetente para atar el mRNA de la ferritina, dejándolo
disponible para la traducción.
Conglomerados De Sulfuro De Hierro
Las proteínas con centros o conglomerados de sulfuro de
hierro llevan a cabo funciones
importantes en diferentes procesos de
transporte de electrones y en reacciones enzimáticos. En
las células
eucariota se conocen proteínas con centro Fe/S en la
mitocondria (aconitasa, subunidades del complejo respiratorio I,
II, III), en el citosol (glutamato sintasa, Isopropil malato
sintasa, Leu I, IRP-1).
Los centros Fe/S, son grupos
prostéticos, los mas comunes encontrados en
proteínas son (2Fe/2S, 4Fe/4S, 3Fe/4S).
En las mitocondrias se produce el ensamblaje de las
proteínas Fe/S endógenas a partir de centros Fe/S,
así como también esta implicada en la
síntesis de centros de Fe/S destinados a proteínas
citosolicas.
Proteínas Participan En La Síntesis De Los
Centros De Fe/S En Humanos
PROTEINA | FUNCION | |
hNFS 1 (mit/cit/nuc) | Cistein desulfurasa | |
IscU1 (cit) IscU2 (mit) | Unen el Hierro al Centro de Fe/S | |
hNFU1 | Maduración del centro Fe/S | |
Adrenodoxina | Participa en reacciones redox | |
Adrenodoxina reductasa | Reduce Yah 1 | |
hISA1 | Incorpora los centros de Fe/S a las apoproteinas | |
hABC7 | Transportador ABC de los centros de Fe/S hacia el | |
Frataxina | Homeostasis del Hierro; biosíntesis de los centros Fe/S | |
BCAT | Exportación del centro Fe/S al citosol | |
(Mit: mitocondrial / cit: citoplasmatico / nuc:
nuclear)
Síntesis Del Grupo
Hem
En las células vivas el HEM se sintetiza por una
vía que ha sido objeto de muchos estudios. Los dos
materiales que
participan son la Succinil CoA, proveniente del ciclo del acido
citrico de las mitocondrias, y el aminoácido Glicina.
En esta reacción también se necesita el fosfato
de piridoxal (vitamina B6), para activar a la Glicina.
Antes de esto, los centros de sulfuro de hierro se exportar de
la mitocondria para formar parte de proteínas y
controlarlas, como la Proteína Reguladora de Hierro Tipo
1, que a su vez regula a otra enzima llamada ALA2
(Aminolevulinato eritroide), que cumple una función
clave en la síntesis del HEM.

Marco
teórico
1. HIERRO
1.1. Antecedentes
El hierro (Fe) es el cuarto elemento más abundante en
la tierra
después del oxígeno, el silicio y el aluminio. Es
un oligoelemento ampliamente distribuido en la naturaleza y
que en el organismo desempeña funciones vitales, puesto
que participa prácticamente en todos los procesos de
óxido-reducción. Este elemento se encuentra
formando parte esencial de las enzimas del ciclo
de Krebs, en la respiración celular y como transportador de
electrones en los citocromos. A su vez, está presente en
numerosas enzimas involucradas en el mantenimiento
de la integridad celular, tales como catalasas, peroxidasas y
oxigenasas.
Este micronutriente cumple un rol esencial en el transporte de
oxígeno, ya que se combina con proteínas para
formar la hemoglobina, forman parte de las células
sanguíneas que transportan oxígeno a los distintos
tejidos del
organismo. También forma parte de la mioglobina que es la
responsable del color rojo de los
músculos y del almacenamiento de
oxígeno por los mismos.
La deficiencia de Fe constituye uno de los trastornos
nutricionales de mayor extensión a
nivel mundial.
Aproximadamente el 20% de las mujeres, el 50% de las mujeres
embarazadas y el 3% de los hombres presentan deficiencia de
hierro. Las causas de deficiencia de hierro son: escaso aporte de
hierro dietético, anomalías en el tracto
gastrointestinal y pérdida de sangre.
1.2. Propiedades químicas del hierro (Fe)
El hierro se puede encontrar en dos formas químicas. En
su forma sólida, existe en forma de metal o en compuestos
que lo contienen. En solución acuosa, el hierro se
encuentra en dos estados de oxidación, Fe2+ (forma
ferrosa), y Fe3+ (forma férrica).
Una propiedad
especial del hierro es su facilidad para cambiar entre estas dos
formas, lo que le permite actuar como catalizador en la
reacciones redox, donando o aceptando electrones. Algunas de las
principales actividades biológicas de las sustancias que
contienen hierro en relación con el oxígeno y con
el metabolismo energético dependen de la propiedad
reactiva del elevado potencial redox. Esta reactividad del hierro
puede ser extremadamente peligrosa, es por esto que se encuentran
estrictamente controladas en el organismo, gracias a la
captación del metal por una proteína transportadora
o por la presencia de otras moléculas con
características antioxidantes.
Hay que agregar que el hierro, en el ambiente
reductivo celular, también tiene efectos dañinos,
pues reacciona no-enzimáticamente con
peróxidos.
1.3. Absorción del Hierro (Fe)
La absorción del hierro ocurre en el
duodeno y yeyuno superior del sistema
gastrointestinal. En el estómago, si bien no se produce la
absorción de este elemento, el mismo contribuye a dicho
proceso, a través de la secreción de ácido
clorhídrico y enzimas, que ayudan no solo a liberar al
hierro de la matriz
alimentaría sino también a solubilizarlo, ya que el
ácido clorhídrico favorece la reducción de
este catión a la forma ferrosa.
El transportador de metales divalentes-1 (DMT1, conocido
anteriormente como Nramp2 o DCT1) es una proteína de
membrana de una sola cadena que tiene 12 regiones
transmembranosas. Interviene en el transporte de hierro desde la
luz intestinal al enterocito y en el resto de las células
interviene en al paso del endosoma al citoplasma.
El DMT1 es una proteína que era conocida anteriormente
por las siglas en inglés Nramp2 (natural resistance
associated macrophage protein) o DCT1( divalent cation
transporter ); es el primer transportador de hierro
caracterizado al nivel molecular en mamíferos. Esta transfiere el hierro a
través de la membrana apical de la célula absortiva
y hacia su interior a través de un proceso acoplado a
protones, por lo que se plantea que actúa en 2 puntos
diferentes: como transportador responsable de la absorción
de hierro en el intestino y en la movilización del mineral
a partir de los endosomas durante el ciclo de la transferrina,
donde transporta el hierro liberado hacia el citoplasma de los
precursores eritroides.
Tiene la singularidad de no ser específico para el
hierro, sino que además transporta desde la luz intestinal
al interior celular otros metales pesados como manganeso,
cobalto, cobre, zinc,
cadmio y plomo. Sin embargo, no transporta calcio ni
magnesio.
La expresión de esta proteína es regulada por
las reservas corporales de hierro, pero también responde
al nivel de hierro dietético, y puede ser controlada por
mecanismos pos-traduccionales , ya que contiene un IRE en su
región 3'UTR, lo que es indicativo de que puede degradarse
en el contexto de un pool de hierro libre elevado, como
ocurre con el mensajero del RTf 1.
1.3.1 Absorción del hierro no-hem
El hierro inorgánico por acción
del ácido clorhídrico del estómago pasa a su
forma reducida, hierro ferroso (Fe 2+), que es la forma química soluble capaz
de atravesar la membrana de la mucosa intestinal. Para absorberse
debe, en una primera etapa, encontrarse en forma soluble, ya que
las formas insolubles no pueden ser absorbidas y son eliminadas
juntamente con las heces
La absorción del hierro no-hem depende de la
solubilidad en la parte alta del intestino delgado, y de
cómo los otros elementos provenientes de la dieta afecta
la solubilidad del metal. También, la absorción del
hierro no-hem es proporcional a la cantidad de inhibidores y
potenciadores de la solubilidad. Dentro de los potenciadores de
la absorción, también llamados agentes reductores
dietarios o Dietary Reducing Agents (DRA), tenemos el
ácido ascórbico, el ácido cítrico,
los cuales forman quelatos de hierro de bajo peso molecular,
facilitando así la absorción del metal en el
duodeno.
Este efecto de los agentes dietarios se ve facilitado por el
citocromo B duodenal. Inversamente, la absorción de hierro
es inhibida por fitatos y taninos. Los fitatos están
presentes principalmente en el trigo y otros cereales, mientras
que los taninos son frecuentes en el té.
La absorción del hierro ocurre principalmente en el
duodeno y parte superior del yeyuno. La membrana de la mucosa
intestinal tiene la facilidad de atrapar al hierro y permitir su
paso al interior de la célula, debido a la existencia de
un receptor específico en la membrana (DMT1).
Una vez que el hierro entra a la célula, la
ceruloplasmina se encarga de oxidar el Fe+2 a Fe+3, para que
así sea captado por la apotransferrina, que se transforma
a transferrina.
El hierro que excede la capacidad de transporte intracelular
es depositado como ferritina, de la cual una parte puede ser
posteriormente liberada a la circulación.

Figura. Mecanismos implicados en el transporte de Fe hacia los
entericitos duodenales.
1.3.2. Absorción del hierro hemo:
Este tipo de hierro atraviesa la membrana celular como una
metaloporfirina intacta, una vez que las proteasas endoluminales
o de la membrana del enterocito hidrolizan la globina. Los
productos de
esta degradación son importantes para el mantenimiento del
hemo en estado
soluble, con lo cual garantizan su disponibilidad para la
absorción. En el citosol del enterocito, la hemoxigenasa
libera el Hierro de la estructura
tetrapirrólica y pasa a la sangre como hierro
inorgánico, aunque una pequeña parte del hemo puede
ser transferido directamente a la sangre portal.
Aunque el hierro hem representa una pequeña
porción del hierro total de la dieta, su absorción
es mucho mayor (20 – 30%) y está menos afectada por
los componentes de DRA: esta. No obstante, al igual que la
absorción del hierro no-hem, la absorción del hem
es favorecida por la presencia de carne en la dieta, posiblemente
por la contribución de ciertos aminoácidos y
péptidos liberados de la digestión, que lo
mantienen soluble y por lo tanto, disponible para la
absorción. Se habla que la absorción del hierro hem
es ayudada o potenciada por un "factor carne", que aún no
esta del todo elucidado. Sin embargo, el ácido
ascórbico tiene poco efecto sobre la absorción del
hemo. Por su parte, el calcio disminuye la absorción de
ambos tipos de hierro por interferir en la transferencia del
metal a partir de la célula de la mucosa, no así en
su entrada a ésta.
1.4. TRANSPORTE DEL HIERRO
1.4.1 Entrada en la circulación
sistémica
En este paso intervienen "exportadores" de hierro como la
hefestina y la ferroportina-1. La hefestina es una
proteína análoga a la ceruloplasmina que se
encuentra en la membrana del enterocito. Se ha localizado en el
brazo largo del cromosoma X (Xq11-12). Los ratones sla
con deficiencia en hefestina padecen anemia
hipocrómica y son capaces de absorber Fe de la luz
intestinal, pero tienen disminuida la excreción al
intestino. La consecuencia de ambos efectos es la
acumulación de Fe en el entericito.
1.4.2. Transporte plasmático
Realizado por la transferrina (TF) que es una
glicoproteína sintetizada en el hígado con dos
lugares de unión para el hierro. Interviene también
en el transporte de Fe en el fluido extracelular. El gen se
encuentra localizado en 3q21. La ausencia de transferrina
detectable en plasma constituye la atransferrinemia.
1.5. METABOLISMO DEL HIERRO
La captación celular del hierro se efectúa
mediante un receptor de transferrina (RTf). La Transferrina tiene
dos sitios de enlace. Los sitios ubicados cerca del C-terminal y
N-terminal tienen gran afinidad por el Fe 3+. Sin embargo, el
N-terminal también podría unirse a otros metales
como: Cr, Cu, Mn, Cd, Zn y
Ni.
El receptor de transferrina es una glucoproteína con un
peso molecular de 180 kDa, que está constituido por dos
subunidades iguales de 95 kDa, cada una de las cuales posee 760
aminoácidos y están unidas por dos puentes
disulfuro. Cada subunidad posee un sitio de unión para la
transferrina. Estos receptores se encuentran anclados en la
membrana a través de un dominio
transmembrana, que actúa como péptido señal
interno (Pomka, 1999).
La concentración de estos receptores es máxima
en los eritroblastos (80 % del total de los receptores del
cuerpo), que son los progenitores nucleados de los
hematíes, donde el hierro es captado por las mitocondrias
para ser incluido en las moléculas de protoporfirina
durante la síntesis del grupo hem.
La afinidad del RTf es sustancialmente mayor para la
transferrina diférrica que para la apotransferrina, siendo
sus constantes de disociación (Kd) de 1,1×10-8 M y
4,6×10-6 M respectivamente. Sin embargo, la concentración
plasmática de transferrina es del orden de 30-40×10-6 M;
esta situación implica que a dicha concentración
los RTf de la superficie celular se encuentran saturados.
Por ello la captación celular del hierro está
regulada por el número de RTf presentes en la superficie,
valor que
dependerá del estado intracelular para el hierro.
Así por ejemplo, aquellos tejidos metabólicamente
activos, donde
aumentan los requerimientos intracelulares de hierro
existirá un mayor número de RTf en la superficie
celular, valor que aumentará ya sea a través de la
síntesis de nuevos RTf o por aumento en la velocidad de
translocación de dicho receptor. De esta manera
aproximadamente 1/3 de la masa total de los RTf está
presente en la superficie de la célula (135-138).
Una vez que la transferrina que posee hierro (TfFe) se une al
RTf en la superficie de la célula, el complejo RTf-TfFe es
captado por la célula por endocitosis. En este proceso la
fracción citoplasmática del receptor juega un rol
esencial en el proceso de internalización del complejo
RTf-TfFe, estando este proceso de internalización regulado
por la activación de la proteína quinasa C. Dentro
del endosoma existe un cambio de
pH a valores
cercanos a 5,5 mediado por una bomba de protones ATP-dependiente,
que produce una disminución de la afinidad de la
transferrina por el Fe.
También existe una unión de Cl- a un sitio de
fijación de aniones del complejo que facilita la
separación del Fe, como así también existe
un proceso reductivo del hierro férrico a su forma
ferrosa, que disminuye aún más la afinidad de la
transferrina por este metal. Este último proceso puede
estar mediado por el ácido ascórbico o
enzimáticamente a través de una enzima endosomal
NADH dependiente. Recientemente se ha demostrado que los grupos
fosfato y pirofosfato también facilitan la
liberación del hierro unido a la transferrina. Este efecto
se ha observado no solo a pH ácido sino también a
pH de 7,4, evidenciando de esta forma un mecanismo secundario de
liberación del hierro del complejo RTf-TfFe. Por otra
parte, se ha observado que la liberación del primer
átomo
de hierro por la transferrina diférrica produce un cambio
en la estabilidad del complejo RTf-TfFe como consecuencia de la
interacción transferrina-receptor que
desestabiliza la unión del átomo de hierro
restante, facilitando de esta manera la liberación del
mismo.

Figura . Ciclo de la Transferrina (Liberona,
2003)
Posteriormente, la fracción del endosoma que contiene
hierro se separa y el hierro de su interior es transferido al
citoplasma de la célula, este proceso aparentemente
podría estar mediado por la bomba de protones
ATP-dependiente. Una vez que el hierro se encuentra en el
citoplasma éste se une a proteínas fijadoras de
hierro o a ligandos de bajo peso molecular. Este hierro,
posteriormente se podrá unir a las proteínas
reguladoras de hierro, integrarse a las estructuras de
las proteínas que poseen hierro o formar parte de los
depósitos celulares de este metal.
La otra parte del endosoma que contiene el complejo apoTf-RTf
se dirige al aparato de Golgi para ser empacado junto a RTf
recién sintetizados. Estas vesículas se dirigen a
la membrana de la célula con la que se fusionan poniendo
en contacto los complejos apoTf-RTf con el espacio extracelular.
A pH del espacio extracelular (7,4) disminuye sustancialmente la
afinidad del RTf por la apoTf y esta última es liberada
para que pueda cumplir nuevamente sus funciones. Este ciclo dura
aproximadamente unos 10 minutos y el mismo puede repetirse unas
100 veces hasta que la transferrina o su receptor sean
degradados.
1.6. ALMACENAMIENTO INTRACELULAR DEL
HIERRO
1.6.1. Ferritina
El hierro se almacena en el citoplasma unido a la
ferritina,consta de 12 dimeros que forman un
dodecaedro de manera que se consigue una doble función:
detoxificación y disponibilidad inmediata de hierro
intracelular.
Cada molécula de ferritina consta de los
monómeros son de tipo H (pesadas) ó
L (ligeras).
La subunidad H tiene un peso molecular de 21.100,
está codificada en un gen localizado en 11q12, y tiene
un sitio de unión para la ceruloplasmina. Está
implicada en la oxidación del hierro ferroso La
ausenciade subunidad H produce la hiperferritinemia con
catarata congénita.La subunidad L tiene un peso de 19.700 está
codificada por un gen que se localiza en 19q13 y no tiene
sitio de unión de la ceruloplasmina. Está
implicada en la formación del núcleo de hierro
de la ferritina
El hígado y el bazo almacenan ferritina. La
ferritina se localiza en prácticamente
todas las células del cuerpo y lípidos
tisulares. La ferritina plasmática correlaciona con
los almacenes
totales del cuerpo por lo que su medida
es Dx en desordenes del metabolismo del Fe.
La ceruloplasmina es una glicoproteína que contiene el
95% del cobre encontrado en el plasma. Se piensa que es la
ferroxidasa que produce el paso de Fe2 + a Fe3 +, y que no tiene
ningún papel directo en el transporte del cobre.
Está codificada por un gen que se encuentra en 3q21-24
cuyas mutaciones dan lugar a la aceruloplasminemia
La biosíntesis de la ferritina es controlada por la
concentración del hierro en la célula.
Interesantemente, este control
está en el nivel de la traducción. Una
proteína conocida como la proteína reguladora del
hierro (IRP) o proteína de unión al elemento de
respuesta al hierro
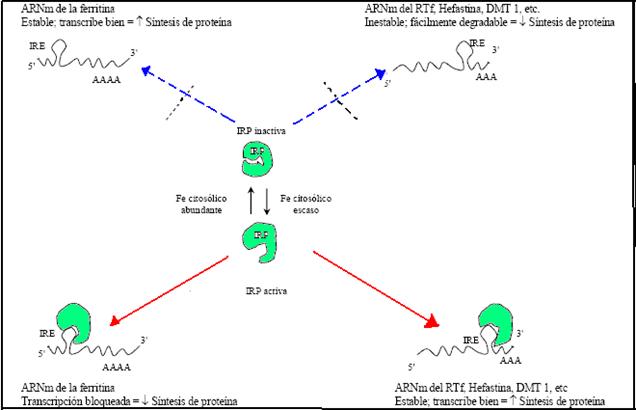
1.7. PROTEÍNA REGULADORAS DEL HIERRO
Con el descubrimiento de las proteínas reguladoras de
hierro (conocidas por sus siglas en inglés, IRP) capaces
de unirse a los elementos de respuesta al hierro o IRE (del
inglés iron responsive elements) de los ARN
mensajeros (ARNm) de las proteínas implicadas en el
metabolismo del mineral, se amplió el horizonte para
comprender los complejos mecanismos del mantenimiento de la
homeostasia del hierro
Asi la disponibilidad de hierro intracelular está
controlada por las proteínas reguladoras de hierro: IRP-1
e IRP-2 que son represores traducionales de la síntesis
del receptor de transferrina, DMT1 y de ferritina.
Esta proteína posee un conglomerado o centro del
sulfuro de hierro (4Fe-4S) que le permite cambiar entre dos
actividades diferentes en dependencia del nivel de hierro
celular.
En el citoplasma, las proteínas IRP se unen a una
región específica de los RNA mensajeros que reciben
el nombre de IRE (elementos con respuesta al hierro). Ambas
proteínas disminuyen la tasa de síntesis de los
genes encargados de la absorción, transporte y
almacenamiento de Fe.
Los IRE son estructuras lazo-tallo localizadas en
las regiones 5' o 3' no traducidas de los ARNm (5' o 3' UTR) que
codifican las proteínas que intervienen en el metabolismo
del hierro.

Fig. 1. Representación
esquemática de la regulación del metabolismo del
hierro. IRE: elemento de respuesta al hierro; IRP:
proteína reguladora de hierro; RTf : receptor de
transferrina : DMT 1: Transportador de metales divalentes
1
Las IRP trabajan en conjunto con estos elementos para
monitorear y responder a los cambios en la cantidad de hierro que
hay en el ambiente intracelular conocido como compartimiento
pool de hierro lábil.
A través de la interacción de las IRPs con los
IREs, la incorporación de hierro vía transferrina
aumenta por estabilización del ARNm del RTf , mientras el
almacenamiento como ferritina disminuye por bloqueo de la
traducción del ARNm de esta proteína. Estos
eventos
resultan en un aumento del pool de hierro lábil.
Inversamente, la incorporación de transferrina disminuye y
el nivel de ferritina aumenta cuando la concentración
intracelular de hierro es elevada
Así, cuando la concentración del hierro es baja,
IRP-1 no es limitado por el hierro y es capaz de atar a extremo
5' del RNA del mensajero de la ferritina en un sitio llamado el
elemento regulador del hierro, un proceso que bloquea la
traducción del mRNA. Inversamente, cuando la
concentración del hierro es alta, y la célula
necesita ferritina, el IRP-1 ata el hierro y llega a ser
incompetente para atar el mRNA de la ferritina, dejándolo
disponible para la traducción (Bowen, 2001).
1.7.1. CONGLOMERADOS DE SULFURO DE HIERRO
a) Generalidades
Las proteínas con centros o conglomerados de sulfuro de
hierro llevan a cabo funciones importantes en diferentes procesos
de transporte de eletrones y en reacciones enzimaticas. Dichas
proteínas están presentes en todos los organismos y
son muy sensibles ha oxidarse. En las células eucariota se
conocen proteínas con centro Fe/S en la mitocondria
(aconitasa, subunidades del complejo respiratorio I, II, III), en
el citosol (glutamato sintasa, Isopropil malato sintasa, Leu I,
IRP-1).
Los centros Fe/S, son grupos prostéticos, los mas
comunes encontrados en proteínas son (2Fe/2S, 4Fe/4S,
3Fe/4S).
La biosíntesis de estos centros no se conoce con
detalle, las primeras investigaciones
de su biosíntesis se los realizo en bacterias y
mas tarde se los observo en células eucariota.
En las mitocondrias se produce el ensamblaje de las
proteínas Fe/S endógenas a partir de centros Fe/S,
así como también esta implicada en la
síntesis de centros de Fe/S destinados a proteínas
citosolicas.
b) Proteínas participan en la síntesis de los
Centros de Fe/S en Humanos
Para la síntesis de centros de Fe/S las mitocondrias
usan un conjunto de proteinas llamadas genericamente complejo ISC
(iron-sulfur cluster assembly) localizadas en diferentes
compartimientos celulares, no solo en las mitocondrias.
Cuadro 1. Proteínas conocidas que
participan en la síntesis de los Centros de Fe/S en
Humanos.
PROTEINA | FUNCION | |
hNFS 1 (mit/cit/nuc) | Cistein desulfurasa | |
IscU1 (cit) IscU2 (mit) | Unen el Hierro al Centro de Fe/S
| |
hNFU1 | Maduración del centro Fe/S | |
Adrenodoxina | Participa en reacciones redox | |
Adrenodoxina reductasa | Reduce Yah 1 | |
hISA1 | Incorpora los centros de Fe/S a las apoproteinas | |
hABC7 | Transportador ABC de los centros de Fe/S hacia el | |
Frataxina | Homeostasis del Hierro; biosíntesis de los | |
BCAT | Exportación del centro Fe/S al citosol | |
Mit: mitocondrial / cit: citoplasmatico /
nuc: nuclear
hNFS1
Proporciona azufre elemental a partir de la cisterna para la
síntesis de Fe/S, esta actividad es tanto mitocondrial,
como citosólico. Es esencial para aconitasa o la
succionato deshidrogenada y disminuyen cuando NFS1 falta o esta
inactiva. También se le designa una función de
capitación y distribución del hierro intracelular.
ISU1- lSU2.
Se encuentran en la matriz mitocondrial, contienen residuos de
cisteína en el centro activo, siendo necesarios para la
función de las proteínas que se unen al hierro.
ISU1 esta localizada en el citosol y en el núcleo, ISU 2
se encentra en la mitocondria.
NFU.
Esta involucrado en la biosíntesis de centros Fe/S,
pero su función bioquímica
aun no se la conoce.
ISA-1
Tienen tres cisteinas que se encuentran conservadas en esta
proteína, son esenciales ya que forman un motivo de
unión a hierro.
Chaperonas
Necesita chaperonas Ssq 1 y Jac 1:
Ssq 1 : Esta involucrada en el metabolismo del hierro, su
ausencia produce acumulación de este metal e la
,mitocondriaJac1: Es una proteína esencial, que podría
dirigir a Ssq1 a sustratos específicos, su
concentración es mas elevada que Ssq1.
ABC
Muchas proteínas Fe/S conocidas son mitocondria les,
pero también las hay localizadas en el citosol. Ejemplos
son la glutamato sintasa, una subunidad de sulfito reductasa, la
isopropil malato y la proteína reguladora de hierro 1
(IRP-1).
La maduración de proteínas con Fe/S
extamitocondriales depende de la maquinaria ISC (iron-sulfur
cluster assembly). Asi NFS1 e ISU1/2 intervienen en la
maduración de proteínas citosolicas solo cuando se
hallan en la mitocondria.
Así los centros Fe/S de proteínas
citosólicas son formados en la matriz mitocondrial y luego
ser exportadas al citosol, existe dos mecanismos para este
proceso:
1) Las apoproteinas podrían ser importadas
desde el citosol a la matriz mitocondrial, donde se
ensamblarían al centro Fe/S para posteriormente ser
exportada al citosol.2) El centro Fe/S formado en la mitocondria
sería exportada al citosol mediante los
transportadores ABC (ATP binding cassette) que comprenden una
familia de proteínas que catalizan el transporte
activo de una amplia variedad de sustratos a través de
la membrana, en este caso hablamos de ABC7
específicamente para los centros de Fe/S.
La ausencia de muchos de los genes de la síntesis de
los centros de Fe/S producen acumulación de hierro
mitocondrial, así como disminución del hierro
citosolico y daño al
DNA mitocondrial.
La acumulación de hierro en la mitocondria produce que
las células sean mas sensibles a oxidarse que produce
defecto e el gen de frataxina (proteína implicada en
mantener la homeostasis
del hierro), causante de Ataxia de Friedreich, enfermedad
neurovegetativa que afecta a una de cada cincuenta mil personas,
en donde hay una deficiencia de ATP y como consecuencia una
alteración en el metabolismo del hierro.
En mamíferos se ha descrito una Proteína
Reguladora de Hierro Tipo I (IRP-1) que tiene un centro 4Fe/4S en
situaciones normales de hierro. Cuando la célula presenta
una concentración de hierro insuficiente el centro de Fe/S
no esta presente en IRP-1 y la apoproteina se une a unas
secuencias de RNA llamadas IRE, que conllevan un aumento en la
expresión de proteinas involucradas en la
asimilación del hierro.
La deficiencia de vitamina E, causa cuadros clínicos
similar a ataxia de Friedreich, seria posible que la ataxia
interviniera en el transporte de hierro al exterior de la
mitocondria y que cooperara con la vitamina E, para proteger a
las células contra estrés
oxidativa.
Estudios han demostrado de estos centros o conglomerados de
sulfuro de hierro en la célula es muy importante para
cotrolar la Proteina Reguladora de Hierro 1 (IRP-1). A su vez
esta IRP-¡ regula a otra enzima llamada ALAS 2 que cumplen
una función clave en la síntesis del HEM.
1.8. SÍNTESIS DEL HEM
1.8.1. ANTECEDENTES
En 1974 Shemin y Rittenberg demostraron que los
nitrógenos del HEM derivan de la Glicina
y que los carbonos derivan de Glicina y Acetato (en forma de
Succinil-CoA).
Las reacciones iniciales son comunes al Hem, la Clorofila y la
Vitamina B12. Grupo prostético de la Hemoglobina y de los
citocromos de las enzimas con Citocromo P-450.
Ocurre en todos los tejidos, principalmente en médula
ósea (Hemoglobina) e hígado (citocromo P450).
1.8.2. SÍNTESIS DEL HEM
Succinil-CoA + Glicina



1.9. PORFIRIAS
Las porfirias (del griego porphuros que
significa púrpura) son un grupo de trastornos
hereditarios o adquiridos debido a deficiencias en la actividad
de enzimas específicas en la vía biosintésis
del HEM. Las deficiencias enzimáticas pueden ser
parciales o casi completas.
En consecuencia, las porfirinas y/o sus precursores son
anormalmente producidos en exceso, se acumulan en los tejidos, y
se excretan en las heces y orina. Estos trastornos se clasifican
en hepáticos y eritropoyéticos, dependiendo del
sitio primario de sobreproducción y acumulación de
las porfirinas o sus precursores.
Las principales manifestaciones de las porfirias
hepáticas son neurológicas (dolor abdominal,
neuropatía y trastornos mentales), mientras que las
porfirias eritropoiéticas característicamente
causan fotosensibilidad cutánea.
Los síntomas de las porfirias son inespecíficos,
y el diagnóstico frecuentemente se retrasa. El
diagnóstico definitivo de la enfermedad radica en la
identificación de la deficiencia enzimática
específica. El aislamiento y caracterización del
ADN que
codifica las enzimas de la biosíntesis
del hem permiten definir las lesiones moleculares responsables
de cada porfiria.
1.9.1. ANEMIA SIDEROBLÁSTICA LIGADA AL CROMOSOMA
X
Las Proteinas Reguladoras de Hierro Tipo 1 se unen a los IRE
de apoferritina para regular la síntesis de
……………….
a) Anemias sideroblásticas
Las anemias sideroblásticas (AS) son enfermedades
metabólicas que tienen una alteración en la
biosíntesis del grupo HEM. No son enfermedades
neoplásicas pero se incluyen en este apartado porque
plantean el diagnóstico diferencial con los SMD.
Las anemias sideroblásticas (ver Tabla) son un conjunto
de enfermedades caracterizadas por una anemia con sideroblastos
en anillo en la médula ósea que son precursores de
los hematíes con gránulos de hemosiderina, que se
tiñen mediante la tinción de Perls, en más
de la tercera parte de la circunferencia del núcleo. Se
ven en otras anemias pero en éstas son muy frecuentes
(más de un 15 por ciento de los sideroblastos son en
anillo). En sangre periférica la anemia es
microcítica en las formas hereditarias ligadas al
cromosoma X y macrocítica en las adquiridas
primarias.
Tabla |
Hereditarias Ligado X (defecto de ALA
|
Anemia sideroblástica hereditaria. Son enfermedades muy
raras. Presenta varias formas de transmisión siendo la
más frecuente la ligada a X, por lo que afecta casi
exclusivamente a varones. ´
Se diagnostican en la infancia o
adolescencia
pero en ocasiones en la edad adulta, porque el grado de anemia es
muy variable. En las mujeres debido a la inactivación del
otro cromosoma X (lionización) pueden en algún
momento tener anemia, pero lo habitual es que sean portadoras sin
anemia, aunque con dos poblaciones de eritrocitos en sangre: una
normocítica y otra microcítica.
En estudios se encontraron con microcitosis con
hematíes con punteado basófilo. El hierro
sérico, la ferritina y la saturación de la
transferrina están elevados; en la médula hay una
hiperplasia eritroide y un aumento del hierro de depósito
y en los eritroblastos (sideroblastos en anillo). El
diagnóstico diferencial es con la hemocromatosis primaria;
en ésta no existe microcitosis y con las anemias
sideroblásticas adquiridas. Responden a piridoxina un
25-50 por ciento. En caso de que no haya respuesta a la B6, el
tratamiento es sustitutivo con transfusiones y en caso necesario
quelante con desferroxiamina.
Otra anemia sideroblástica hereditaria es el
síndrome de Pearson, enfermedad muy grave que tiene una
anemia sideroblástica macrocítica asociado a
disfunción pancreática exocrina, hepática y
renal.
Anemias sideroblásticas adquiridas. La forma primaria
es un tipo de síndrome mielodisplásico que cursa
con macrocitosis. Las formas adquiridas secundarias son debidas
a:
Alcohol: asociado a malnutrición y déficit
de fólico, se produce una anemia sideroblástica
con mejoría al suspender el alcohol, dar piridoxina
temporalmente y seguir una dieta equilibrada.Isoniazida: es un antagonista de la vitamina B6 y produce
una anemia sideroblástica microcítica
después de 1-10 meses de tratamiento en personas
predispuestas. Desaparece al retirar el fármaco o
administrar simultáneamente dosis bajas de
B6.Cloramfenicol: es un efecto predecible (no
idiosincrásico como la anemia aplásica) de su
uso a dosis altas. Es reversible al retirar el
medicamento.Toxicidad por plomo o saturnismo: en la médula
ósea pueden verse sideroblastos en anillo, pero la
anemia es multifactorial y predominantemente
hemolítica. El plomo inhibe la síntesis del
grupo hemo y de las cadenas globínicas de la Hb en
varios puntos.
b) Anemia sideroblástica ligada al cromosoma
X
Aunque existen casos sin aparente afectación familiar
previa, la mayoría de las anemias sideroblásticas
tienen un patrón hereditario. Las más frecuentes
presentan herencia ligada
al sexo y afectan
fundamentalmente a los varones, aunque las mujeres portadoras
puedan excepcionalmente sufrir anemia leve o rasgos
hematológicos característicos de la enfermedad. Se
han descrito pocos casos bien documentados de anemia
sideroblástica constitucional de herencia autosómica, dominante o recesiva.
El trastorno enzimático mejor caracterizado en estas
anemias congénitas es el déficit de ALA-sintasa
(delta-aminolevulinato sintasa), sobre todo en las formas
hereditarias ligadas al cromosoma X. Este déficit se debe
a mutaciones puntuales del gen ALAS2 localizado en Xp11.21. Los
individuos afectados desarrollan anemia en la infancia y muerte por
sobrecarga férrica al cabo de pocos años.
Otra forma de anemia sideroblástica hereditaria es la
ligada al cromosoma X con ataxia (XLSA/A). Se caracteriza por una
ataxia espinocerebelosa no progresiva que se manifiesta en los
primeros años de vida. La mutación responsable
afecta a un gen localizado en Xq13 que codifica la
proteína ABC7 (ATP-binding cassette 7), que está
implicada en el transporte del grupo HEM, desde la mitocondria.
Las mutaciones en ABC7 producen una acumulación de hierro
en la mitocondria que afecta principalmente a las células
neuronales y eritroideas.
Metabolismo mitocondrial del Fe
Se ha sugerido la existencia de un ciclo mitocondrial del
hierro que implicaría la existencia de un proceso regulado
de entrada, control del almacenamiento de Fe, y un mecanismo de
excreción del Fe mitocondrial (18). Hasta el momento solo
se conocen dos proteínas implicadas en este
proceso:
La frataxina es una proteína pequeña, codificada
por un gen localizado en 9q13, que se encuentra deficiente en los
pacientes con ataxia de Friedreich. El daño neuronal y
cardiaco de esta enfermedad parece estar causado por el
acúmulo de Fe en la mitocondria, por lo que se piensa que
la frataxina tiene una función en la excreción
mitocondrial de Fe.
En la anemia sideroblástica con ataxia ligada al
cromosoma X (XLSA/A), se encuentran mutaciones de la
proteína ABC 7 (ATP-Binding Cassette 7),
relacionada con el transporte del grupo Hemo. Las alteraciones
eritroides y neuronales de estos pacientes parecen tener su
origen en el acúmulo de Fe mitocondrial.
c) Sobrecargas Férricas
Hiperferritinemia con catarata congénita
autosómica dominante
La hiperferritinemia con catarata congénita
autosómica dominante, es una enfermedad del metabolismo
del hierro que se caracteriza por catarata bilateral precoz y
concentraciones elevadas de ferritina sérica (14). La
ferritina se encuentra formada por 24 subunidades de al menos dos
tipos L y H. En individuos normales hay una mezcla de ambos
tipos, mientras que en los afectados por esta enfermedad la
subunidad H es indetectable. Aparte de la aparición de
cataratas, no hay otras manifestaciones clínicas ni
alteraciones hematológicas o bioquímicas.
Los estudios moleculares han identificado múltiples
mutaciones puntuales en el elemento IRE (elemento de respuesta al
hierro) del RNA mensajero de la L-ferritina, afectando al motivo
CAGUGU que forma parte del bucle IRE y que interviene en la
unión de IRP (proteína reguladora del hierro).
Estas mutaciones impedirían la unión de IRP
provocando una síntesis constitutiva y sin
regulación de la L-ferritina.
Ataxia de Friedreich
La ataxia de Friedrich es la ataxia espinocerebelosa
hereditaria más frecuente, tiene un patrón de
aparición autosómico recesivo y se caracteriza por
una degeneración progresiva que afecta al sistema nervioso
central, periférico, y corazón.
El origen de la enfermedad se encuentra en la carencia de
frataxina, una proteína mitocondrial que regula la
expulsión de hierro en la mitocondria. La
acumulación de hierro mitocondrial provoca un
estrés oxidativo y fallos en la cadena respiratoria que
causan daño celular.
Hallazgos Clínicos
Los varones presentan anemia hemolítica refractaria,
palidez y debilidad durante la lactancia.
Presentan hiperesplenismo secundario, sobrecarga de hierro, y
tienen la posibilidad de desarrollar hemosiderosis. La anemia es
microcítica e hipocrómica con notable anisocitosis,
poiquilocitosis y policromasia. Los leucocitos y plaquetas son
normales.
DIAGNÓSTICO
La exploración de la médula ósea muestra
hipercelularidad, con desviación a la izquierda y
eritropoiesis megaloblástica, con maduración
anormal. El diagnóstico definitivo descansa en la
demostración de la deficiencia de ALAS.
TRATAMIENTO
La anemia grave responde a la
administración de piridoxina. Dicho cofactor puede
obviar la necesidad de transfusiones o reducir su frecuencia.
Aquéllos que no responden a la piridoxina dependen de las
transfusiones y requieren quelantes.
Diseño
metodológico
El presente trabajo es un estudio descriptivo y
analítico.
Discusión
Esta revisión recoge las principales
características de la participación de hierro,
así como sus propiedades, efectos fisiológicos y
aplicaciones terapéuticas
Como es de saber el hierro (Fe) es el cuarto elemento
más abundante en la tierra después del
oxígeno, el silicio y el aluminio. Es un oligoelemento
ampliamente distribuido en la naturaleza y que en el organismo
desempeña funciones vitales, puesto que participa en todos
los procesos de óxido-reducción y un rol muy
importante en la formación de algunas proteínas
como la hemoglobina y mioglobina por ejemplo.
La deficiencia de Fe constituye uno de los trastornos
nutricionales de mayor extensión a nivel mundial.
Una de las causas de la aparición de enfermedades
involucradas en alteraciones de la formación del grupo
hemo radica en el papel bioquímico que cumple la
absorción de hierro.
Una vez atravesado la membrana celular como una
metaloporfirina intacta, las proteasas endoluminales o de la
membrana del enterocito hidrolizan la globina. Los productos de
esta degradación son importantes para el mantenimiento del
hemo en estado soluble, con lo cual garantizan su disponibilidad
para la absorción. En el citosol del enterocito, la
hemoxigenasa libera el Hierro de la estructura
tetrapirrólica y pasa a la sangre como hierro
inorgánico, aunque una pequeña parte del hemo puede
ser transferido directamente a la sangre portal.
El hierro se almacena en el citoplasma unido a la ferritina,
que consta de dos monómeros de
tipo H (pesadas) ó L (ligeras), formando
un dodecaedro de manera que se consigue una doble función:
detoxificación y disponibilidad inmediata de hierro
intracelular.
Así la disponibilidad de hierro intracelular
está controlada por las proteínas reguladoras de
hierro: denominadas IRP-1 e IRP-2 que son represores
traduccionales de la síntesis del receptor de
transferrina, DMT1 y de ferritina.
Esta proteína posee un conglomerado o centro del
sulfuro de hierro (4Fe-4S) que le permite cambiar entre dos
actividades diferentes en dependencia del nivel de hierro
celular.
En el citoplasma, las proteínas IRP se unen a una
región específica de los RNA mensajeros que reciben
el nombre de IRE (elementos con respuesta al hierro). Ambas
proteínas disminuyen la tasa de síntesis de los
genes encargados de la absorción, transporte y
almacenamiento de Fe.
Las IRP1 regulan a otra enzima llamada ALAS2 que
desempeñe un papel dominante en síntesis del grupo
hemo. Las IRP trabajan en conjunto con estos elementos para
monitorear y responder a los cambios en la cantidad de hierro que
hay en el ambiente intracelular conocido como compartimiento
pool de hierro lábil
Con lo cual al producir alteraciones en el proceso que
involucran el hierro (absorción, almacenamiento, etc.)
determinan alteraciones en la formación de la hemoglobina
lo cual con lleva a la adquisición de la anemia
sideroblastica.
La anemia sideroblastica una de las enfermedades responsables
de un índice elevados de la mortalidad de niños y
de personas en el mundo, es ocasionada por una síntesis
deficiente del hemo A, la parte de la hemoglobina que contiene
hierro. Esta síntesis se lleva a cabo en los precursores
eritroides e incluye al menos 4 reacciones enzimáticas
intramitocondriales. Cualquier trastorno que afecte uno de estos
pasos impide la incorporación de hierro al hemo A,
originando un depósito de hierro en la mitocondria y un
sideroblasto patológico que muere antes de llegar a
eritrocito.
Cuando la concentración del hierro es baja, IRP-1 no es
limitado por el hierro y es capaz de atar a extremo 5' del RNA
del mensajero de la ferritina en un sitio llamado el elemento
regulador del hierro, un proceso que bloquea la traducción
del mRNA. Inversamente, cuando la concentración del hierro
es alta, y la célula necesita ferritina, el IRP-1 ata el
hierro y llega a ser incompetente para atar el mRNA de la
ferritina, dejándolo disponible para la
traducción
Conclusiones y
recomendaciones
El papel del conglomerado o el centro de sulfuro de hierro son
fundamentales para la formación del grupo hemo. No se han
descrito trabajos de la relación de alteración de
formación del grupo hemo, ni tampoco exclusivos de los
conglomerados de sulfuro de hierro. Es importante resaltar que,
en muchos casos, las alteraciones de la formación del
grupo hemo es por causa de elementos genéticos pero aun es
mas alarmante los altos índices de la adquisición
de la enfermedad.
En este caso el problema se vuelve critico, porque
generalmente se reflejan en alteraciones en el organismo, tal es
el caso de la anemia sideroblastica.
La emergencia de la anemia sideroblastica es una de las
enfermedades de causa considerable de mortalidad de niños
y de adultos. Es bien conocida las alteraciones que produce la
anemia sideroblasticas en el organismo. Como ejemplo de esto se
cuenta las diferentes alteraciones de la formación del
grupo hemo, la de la hemoglobina, la cual se resumen a una
alteración que impide la incorporación de hierro al
hemo A, originando un depósito de hierro en la mitocondria
y un sideroblasto patológico que muere antes de llegar a
eritrocito.
La única alternativa que se vislumbra para evitar una
progresión de esta situación consiste en aplicar
programas
efectivos que vinculen charlas en la importancia del hierro en la
nutrición,
y el de apoyar todo trabajo de investigación que se realice en post de
encontrar alguna solución para contrarrestar la anemia
sideroblastica y de esa forma disminuir el índice de
mortalidad de niños y de adultos en el mundo.
Bibliografía
1.- Simon M, Bourel M, Genetet B. and Fauchet R. Idiophatic
hemochromatosis: demonstration of recessive transmission and
early detection by family HLA typing. N Engl J Med 1977; 297:
1017-1021.
2. Feder JN, Gnirke A, Thomas W, et al. A novel MHC class-I
like gene is mutated in patients with hereditary
haemochromatosis. Nat Genet 1996; 13: 399-408.
3. Andrews NC, Fleming MD, Levy JE, Molecular insights into
mechanisms of iron transport. Curr Opin Hematol 1999; 6:
61-64
4. Vulpe CD, Kuo YM, Murphy TL. Hephaestine, a ceruloplasmin
homologue implicated in intestinal iron. transport, is defective
in the sla mouse. Nat.
Genet. 1999; 21: 195-199.
5. Ponka P, Beaumont C, Richardson DR, Function and regulation
of transferrin and ferritin. Semin Hematol 1998; 35:
35-54.
6. Parkkila S, Waheed A, Britton RS, et al. Association of the
transferrin receptor in human placenta with HFE, the protein
defective in hemochromatosis. Proc Natl Acad Sci USA 1997; 94:
13198-13202.
7. Murray R , Mayes P , Granner D , Rodwell V .
Bioquímica de Harper, 15ª ed., Mexico-D.F. ; Edit
EL MANUAL MODERNO : 20047. 8. Zon L et al. Developmental Regulation of
Hematopoiesis / publicación periódica en
línea / 2005 / fecha de consulta 2007 Marzo 22 / 4:2
pantallas. Disponible en:
http://www.hhmi.org/news/zon4-esp.html7. 9. HHMI (Howard Hugles medical Institute) .Los
investigadores descubren la ruta nueva a la síntesis
de hemoglobina /publicación periódica en
línea / 2005 / fecha de consulta 2007 Marzo 22 / 5 :5
pantallas. Disponible en:
http://www.hhmi.org/research/investigators/zon.htmml
Anexos
CONGLOMERADOS DE SULFURO EN LA SINTESIS DEL
HEM
CICLO DE LA TRANSFERRINA
PROTEINA REGULADORA DE HIERRO TIPO 1
CONGLOMERADOS SULFURO DE HIERRO
SINTESIS DEL HEM
CATABOLISMO DEL HEM
ANEMIA SIDEROBLASTICA LIGADA AL CROMOSOMA
X
METABOLISMO DEL HIERRO
MOLÉCULA DE HEMOGLOBINA
ESTRUCTURA DEL GRUPO HEM
SÍNTESIS DE LA HEMOGLOBINA
EL OXÍGENO UNIDO A LA
HEMO-PROTEÍNA
LOCALIZACIÓN DEL HIERRO
CONGLOMERADOS DE SULFURO EN LA SINTESIS DEL
HEM



CICLO DE LA TRANSFERRINA

PROTEINA REGULADORA DE HIERRO TIPO 1

CONGLOMERADOS SULFURO DE HIERRO
PROTEINA | FUNCION | |
hNFS 1 (mit/cit/nuc) | Cistein desulfurasa | |
IscU1 (cit) IscU2 (mit) | Unen el Hierro al Centro de Fe/S
| |
hNFU1 | Maduración del centro Fe/S | |
Adrenodoxina | Participa en reacciones redox | |
Adrenodoxina reductasa | Reduce Yah 1 | |
hISA1 | Incorpora los centros de Fe/S a las apoproteinas | |
hABC7 | Transportador ABC de los centros de Fe/S hacia el | |
Frataxina | Homeostasis del Hierro; biosíntesis de los | |
BCAT | Exportación del centro Fe/S al citosol | |
SINTESIS DEL HEM



CATABOLISMO DEL HEM

ANEMIA SIDEROBLASTICA LIGADA AL CROMOSOMA
X
Anemias Siderobásticas |
Secundaria: alcohol, |
METABOLISMO DEL HIERRO

EL OXIGENO NO
"VIAJA" SUELTO DENTRO DE LOS GLÓBULOS ROJOS SINO
"ENGANCHADO" A UNA PROTEÍNA QUE SE LLAMA HEMOGLOBINA
(HEM)

MOLÉCULA DE HEMOGLOBINA
Esta proteína esta formada por:
monómeros alfa y beta
y en el centro presenta un átomo de hierro.
El hierro (Fe) es un metal que puede estar en dos formas,
oxidada (Fe3+) o reducida (Fe2+). Para poder transportar el
oxigeno, ese átomo de hierro tiene que estar en la
forma reducida.

ESTRUCTURA DEL GRUPO HEM

SÍNTESIS DE LA HEMOGLOBINA


EL OXÍGENO UNIDO A LA
HEMO-PROTEÍNA:
la interacción del Oxígeno
con un grupo distal (parte superior de la figura)y con un grupo proximal (parte inferior de
la figura).La hemoglobina es una cromoproteina compuesta por una
proteína, la globina, unida a una molécula muy
parecida a la clorofila, pero que, en vez de magnesio,
contiene hierro; el oxigeno se le une en forma reversible.
Cuando la hemoglobina esta unida a oxigeno se llama
oxihemoglobina y cuando lo ha soltado
deoxihemoglobina.

El Hierro necesario para la formación de
hemoglobina el ser humano lo toma en su dieta a razón
de 1 miligramo por día, acumulándose
normalmente 4 gramos de el en los adultos. Es decir, un ser
humano adulto tendría hierro suficiente como para
elaborar un clavo de 4 centímetros de largo.
LOCALIZACIÓN DEL HIERRO

AGRADECIMIENTO
Los autores agradecemos a Dios por darnos la vida, a nuestros
padres por inculcarnos con su ejemplo de personas responsabilidad, empeño y
superación, a los autores del material de consulta ya que
con dichos conocimientos publicados fue posible la
realización de este trabajo, a nuestra auxiliar de
docencia
universitaria Carolina Rodríguez que con su
dedicación en la orientación de la referencia
bibliografica fue determinante para la realización del
trabajo , a los docentes de la
cátedra de bioquímica por la iniciativa de la
realización de la elaboración del trabajo.
Autor:
Alcón Mamani Sandra Gladys
Aliaga Gutierrez Juan Victor
Añawaya Rojas Telma
Cahune Chavez Sarela
Fecha: Octubre de 2007
La Paz, Bolivia
UNIVERSIDAD MAYOR DE SAN ANDRES
FACULTAD DE MEDICINA,
ENFERMERIA, NUTRICIÓN Y TECNOLOGÍA
MEDICA
CARRERA DE MEDICINA
DEPARTAMENTO DE BIOQUIMICA Y BIOLOGIA CELULAR

 Página anterior Página anterior | Volver al principio del trabajo | Página siguiente  |