Cuadro Nº1.
Clasificación de las malformaciones
anorrectales
Género | Malformación | Necesidad de colostomía |
Masculino | Fístula cutánea (fístula perineal) | No |
Fístula rectouretral | ||
Bulbar | Sí | |
Prostética | Sí | |
Fístula rectovesical | Sí | |
Agenesia anorrectal sin fístula | Sí | |
Atresia rectal | Sí | |
Femenino | Fístula cutánea (perineal) | No |
Fístula Vestibular | Si | |
Agenesia anorrectal sin fístula | Si | |
Altresia rectal | Si | |
Cloaca persistente | Si |
ANO IMPERFORADO
Y MALFORMACIONES CLOACALES
Incidencia, tipos de defectos y
terminología
El ano imperforado se presenta en uno de cada 4 000 a 5
000 recién nacidos. El riesgo estimado de que una pareja
tenga un segundo hijo con alguna malformación ano
rectal se acerca al 1%. La frecuencia de este defecto es un poco
más alta en varones que en mujeres. El defecto más
común en los varones es el ano imperforado con fístula
rectouretral. La anormalidad más frecuente en mujeres
es la fístula rectoves-tibular. El ano imperforado sin una
fístula es más bien un defecto infrecuente y se
presenta aproximadamente en 5% de todo el grupo de malformaciones. La
cloaca persistente se considera un defecto inusual. En
cambio, en la bibliografía se refiere una alta
incidencia de fístula rectovaginal. En retrospectiva, parece
que la presencia de una cloaca es un defecto mucho más
frecuente en las pacientes femeninas. Además, la
presencia de una cloaca tal vez sea el tercer defecto
más frecuente en las mujeres después de las
fístulas perineales y las vestibulares. De hecho, la
fístula rectovaginal casi siempre es un defecto inexistente
que se presenta en menos del 1% de todos los casos. Es probable
que en la mayoría de los pacientes que sufren por
persistencia de la cloaca se considerara más bien una
fístula rectovaginal. Muchos de esos sujetos se
sometieron a intervención quirúrgica y se
reparó el componente rectal de la malformación,
pero los pacientes quedaron con un trayecto urogenital
persistente. Además, la evidencia reciente muestra que la mayor parte de las
fístulas rectovestibulares se denominó a menudo como
"fístula rectovaginal". La fístula entre el recto y el
cuello vesical en los varones es la única malformación
real superior al elevador y por fortuna sólo ocurre en cerca
del 10% de estas personas. Es el único defecto en los
pacientes masculinos que requiere una laparotomía
además del abordaje inferior para la
reparación.
DEFECTOS
MASCULINOS
FÍSTULAS
RECTOURETRALES
Si los pliegues de Rathke no se forman, la parte
inferior del tabique urorrectal no se cerrará, con la
consiguiente aparición de fistulas rectouretrales
anómalas entre el seno urogenital y el recto en desarrollo. (Larsen,
2003)
En los varones, estas comunicaciones suelen adoptar
la forma de una estrecha fístula uretral
rectoprostática. (Larsen, 2003)
El orificio de la fístula rectouretral, el defecto
más frecuente en el sexo masculino, se puede
localizar en la parte inferior de la uretra (uretra bulbar; fig.
IB) o en la uretra superior (uretra prostética; fig.
1A).

Espectro de malformaciones en pacientes del
sexo masculino. A,
fístula perineal. B, fístula
rectouretral bulbar. (Tomado de Peña A.: Atlas
of Surgical Management of Anorectal Malformations.
New York, Springer-Verlag, 1990, p. 26. Lois Barnes,
dibujante médico.)
Inmediatamente por arriba del sitio de la fístula,
el recto y la uretra comparten una pared común. Este
importante hecho anatómico tiene implicaciones
técnicas y quirúrgicas significativas. Por lo
regular, el recto está distendido y rodeado lateral y
posteriormente por el músculo elevador. Entre el recto y la
piel perineal existe una
porción de músculo voluntario estriado, llamado
complejo muscular. La contracción de las fibras de este
músculo eleva la piel de la depresión anal. A nivel de
la piel, a ambos lados de la línea media, se localiza
un grupo de fibras de músculo voluntario, conocidas
como fibras parasagitales. Por lo general, las
fístulas uretrales bajas (bulbares) se relacionan con
músculos de buena calidad, sacro bien
desarrollado, surco en la línea medía y
depresión anal prominentes. Con mayor frecuencia las
fístulas uretrales altas (prostéticas) se vinculan con
músculos de pobre calidad, sacro anormalmente
desarrollado y perineo plano, con un pobre surco en la
línea media y sin depresión anal visible. No
obstante, existen excepciones para estas reglas. Muchas veces los
pacientes expulsan meconio a través de la uretra, un signo
inequívoco de fístula rectouretral. (Campbell,
2004)
FÍSTULAS
RECTOVESICALES
Esta alteración más grave se produce por un
falta de formación del pliegue de Tourneux y del pliegue de
Rathke. (Larsen, 2003)
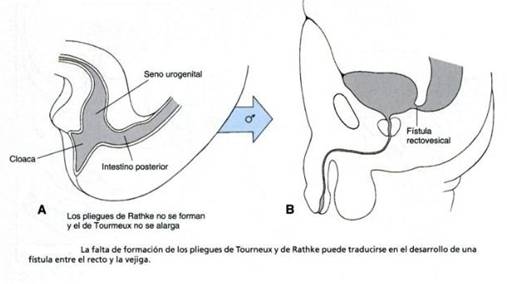
En este defecto, el recto se abre a nivel del cuello
vesical. El sujeto tiene un mal pronóstico, ya que el
músculo elevador, el complejo muscular y el esfínter
externo suelen estar mal desarrollados. Muchas veces el sacro
está deforme. Toda la pelvis parece estar subdesarrollada.
En muchas ocasiones el perineo es plano, con datos de desarrollo muscular
deficiente. Cerca de 10% de los casos de ano imperforado
pertenece a esta categoría.

Espectro de defectos en pacientes masculinos.
A, fístula rectouretral
prostética. 8, fístula recto-cuello
vesical. (Tomado de Peña A.:
Atlas of Surgical Management of Anorectal
Malformations. New York, Spriñger-Verlag, 1990,
p. 26. Lois Barnes, dibujante
médico.)
AGENESIA
ANORRECTAL SIN FÍSTULAS
Es interesante advertir que la mayoría de los
pacientes con este defecto poco común tiene un sacro
bien desarrollado y músculos adecuados. El recto termina
aproximadamente a 2 cm de la piel perineal. Es común que el
individuo tenga un buen
pronóstico en términos de función intestinal.
Incluso cuando el enfermo no tiene comunicación entre el
recto y la uretra, estas dos estructuras están
separadas únicamente por una pared común y delgada, que
es un detalle anatómico relevante con implicaciones
técnicas.
Casi la mitad de las personas sin fístula
también padece síndrome de Down y más del 90% de
los pacientes con este síndrome y ano imperforado muestra
este defecto específico. El hecho de que estos
pacientes tengan síndrome de Down no parece interferir con
el buen pronóstico en términos de control intestinal para esta
malformación.
ATRESIA
RECTAL
En esta anomalía extremadamente rara de los
varones, la luz del recto puede estar
interrumpida en su totalidad (atresia) o de manera parcial
(estenosis). El saco ciego superior está representado por un
recto dilatado, en tanto que la porción inferior
está representada por un conducto anal pequeño,
que mide alrededor de 1 a 2 cm de profundidad. Las estructuras
pueden estar separadas por una membrana delgada o una
porción densa de tejido fibroso. Estos defectos se
observan en cerca de 1% del grupo entero de malformaciones. Los
sujetos con este defecto tienen todos los elementos necesarios
para la continencia y un pronóstico excelente. Debido
a que poseen un conducto anal bien desarrollado, su
sensibilidad en el ano recto es normal. Están presentes
estructuras musculares voluntarias casi
normales.
FÍSTULAS
CUTÁNEAS
La fístula cutánea es un defecto bajo. El
recto se localiza dentro de la mayor parte del mecanismo
del esfínter. Sólo la parte más inferior del
recto está mal colocada en sentido anterior. En
ocasiones, la fístula no se abre hacía el perineo, sino
más bien sigue una vía en la línea media
subepitelial, abriéndose en algún pimío a lo largo
del rafe perineal de la línea media, escroto o incluso la
base del pene. El diagnóstico se establece mediante
inspección perineal. No se requieren investigaciones
adicionales.
Las más de las veces la abertura (fístula)
anal es demasiado estrecha (estenosis). Los términos ano
cubierto, membrana anal y malformaciones en asa de cubo se
refieren a diferentes manifestaciones externas de las
fístulas perineales.
DEFECTOS EN EL SEXO
FEMENINO
FÍSTULAS RECTOURETRALES
Se produce por la falta de desarrollo de los pliegues de
Rathke. Esta situación se complica en el caso de las
mujeres, por la presencia de conductos paramesonéfricos. Lo
más frecuente es que estos conductos se fijen a la uretra
pélvica inmediatamente por encima de la fistula
rectouretral. La región no dividida de la cloaca se
convierte entonces en una desembocadura común para la
vagina, el recto y la uretra, recibiendo el nombre de canal
rectocloacal. En ocasiones la fistula recouretral se incorpora al
canal uterovaginal que desciende en su emigración hacia una
posición más inferior en la pared posterior de la
cloaca. En estos casos, la vagina y la uretra se abren por
separado en el vestíbulo, pero el recto comunica con la
vagina a través de una fistula rectovaginal. La fistula
puede afectar a una porción alta o baja de la vagina. Si la
fistula rectouretral se localiza inicialmente en la unión
entre la vagina y la cloaca, la fistula anovestibular resultante
se abrirá en el vestíbulo
vaginal.
FÍSTULAS
RECTOVESICALES
Esta alteración más grave se produce por un
falta de formación del pliegue de Tourneux y del pliegue de
Rathke. En las mujeres, esta anomalía puede interferir con
la función normal de los extremos inferiores de los
conductos paramesonéfricos, con formación de dos
vaginas y úteros separados que se abren directamente en la
vejiga. (Larsen, 2003)
FÍSTULAS CUTÁNEAS
(PERINEALES)
Desde los puntos de vista terapéuticos y
pronósticos, esta anormalidad común es equivalente a la
fístula cutánea descrita en los varones. El recto
está bien localizado dentro del mecanismo del
esfínter, excepto por su porción más baja, que
está situada anteriormente. El recto y la vagina
están bien separados.
FÍSTULAS VESTIBULARES
En este defecto importante, las pacientes
pediátricas tienen buen pronóstico en términos de
función intestinal, cuando se tratan de manera
apropiada. No obstante, en la experiencia del autor, la
persona es enviada a menudo de
otras instituciones debido a
reparaciones infructuosas. El intestino se abre justo detrás
del himen en el vestíbulo de los genitales femeninos.
Inmediatamente por arriba del sitio de la fístula, el recto
y la vagina están separados por una pared común
delgada. Estas mujeres tienen por lo regular músculos bien
desarrollados y un sacro y nervios normales. Sin embargo, algunas
veces este defecto se acompaña de un sacro mal desarrollado.
(Campbell, 2004)
Con frecuencia, las pacientes con fístula
vestibular son referidas al cirujano con un diagnóstico
erróneo de fístula rectovaginal. El diagnóstico
preciso es clínico. Se requiere una inspección
minuciosa de los genitales de la recién nacida para el
diagnóstico. Algunos cirujanos pediatras reparan este
defecto sin una colostomía de protección. Muchas de
estas personas se recuperan de modo satisfactorio. Empero, una
infección perineal seguida de dehiscencia de la anastomosis
anal y recurrencia de la fístula provoca fibrosis grave que
puede interferir con el mecanismo del esfínter.
En este caso, la paciente podría perder la mejor
oportunidad para un resultado funcional óptimo, ya que las
operaciones secundarias no
proporcionan el mismo buen pronóstico que una
intervención primaria con éxito. Por
consiguiente, se recomienda enfáticamente una
colostomía protectora, seguida por una forma de procedimiento sagital posterior
limitado como reparación final.
El término fístula vaginal se emplea muchas
veces en forma equivocada para referirse a sujetos que en
realidad tienen una fístula vestibular o una cloaca. La
fístula vaginal verdadera se encuentra en menos del 1% de
todos los casos, por lo que no se considera parte de la
clasificación propuesta aquí. (Campbell,
2004)

Espectro de malformaciones en pacientes
femeninas. A, fistula perineal. B,
fistula vestibular. (Tomado de Peña A.: Atlas of
Surgical Management of Anorectal Malformations. New
York, Springer Verlag, 1990, p. 50.' Lois Barnes,
dibujante médico.)
AGENESIA
ANORRECTAL SIN FÍSTULA
Este defecto del sexo femenino tiene las mismas
implicaciones terapéuticas y pronosticas mencionadas para
los varones.
CLOACA
PERSISTENTE
En este grupo de anomalías está representada
la extrema complejidad de las malformaciones femeninas. Una
cloaca persistente se define como una anormalidad en la
cual el recto, la vagina y el aparato urinario convergen y se
fusionan en un solo conducto común.

Espectro de cloacas. A, tipo
más común de cloaca. B, conducto
común largo. (Tomado de Peña A.: Atlas of
Surgical Management of Anorectal Malformations. New York,
Springer-Verlag, 1990, p. 60. Lois Barnes, dibujante
médico.)
El diagnóstico de cloaca persistente es
clínico. Este defecto se debe sospechar en un lactante
femenino con ano imperforado y genitales de aspecto
pequeño. La separación cuidadosa de los labios
revela un orificio perineal único. La longitud del
conducto común varía de 1 a 7 cm, lo cual tiene
implicaciones técnicas y pronosticas. Los conductos
comunes mayores de 3.5 cm representan por lo general un defecto
complejo.
Es difícil la movilización de la vagina. En
consecuencia, con frecuencia se utiliza alguna forma de
sustitución vaginal durante la reparación. Un
conducto común menor de 3.4 cm significa habitualmente que
el defecto se puede reparar con una operación sagital
posterior única, sin abrir el abdomen. En ocasiones, el
recto se abre alto en la cúpula de la vagina. Por lo tanto,
una laparotomía debe ser parte del procedimiento para
movilizar el intestino. Es común asimismo que la
vagina esté distendida de manera anormal y llena de
secreciones mucosas (hidrocolpos). Esta vagina
distendida comprime el trígono y por ende suele
acompañarse de megauréteres. Una vagina grande
representa una ventaja técnica para la reparación, en
virtud de que más tejido vaginal facilita su
reconstrucción. Un hallazgo frecuente de las malformaciones
cloacales es el grado variable de tabicación o
duplicación vaginal y uterina. Por lo regular, el recto se
abre entre las vaginas. Las malformaciones cloacales bajas casi
siempre se vinculan con un sacro bien desarrollado, un perineo de
aspecto normal y músculos y nervios adecuados. Esto
hace posible un buen pronóstico.

Espectro de cloacas. A,
implantación rectal alta en la vagina. B,
conducto común corto. (Tomado do Peña A.: Atlas of
Surgical Management of Anorectal Malformations. New
York, Springer-Verlag, 1990, p. 61. Lois Barnes,
dibujante médico.)
EXTROFIA
CLOACAL
La extrofia cloacal, una de las más grandes
anomalías congénitas compatibles con la viabilidad
intrauterina, es sumamente rara y se produce en 1 de cada 200,000
a 400,000 nacidos vivos (Hurwitz y col., 1987). Los
reportes más recientes indican una relación hombre – mujer de 2:1 (Gearhart y Jeffs,
1998). La herencia de la extrofia cloacal
se desconoce porque nunca se ha sabido de descendencia en
pacientes con este
trastorno.
Anatomía
La extrofia cloacal, también conocida como fisura
vesicointestinal, cloaca ectópica, ectopia visceral,
extrofia complicada de la vejiga y fisura de la pared
abdominal, es probablemente la forma más grave de los
defectos de la pared abdominal ventral.
Los defectos típicos de la extrofia cloacal
consisten en un onfalocele por arriba y una exposición de
intestino y vejiga por abajo. La vejiga es bivalva en la
línea media por una zona de mucosa intestinal y cada
hemivejiga contiene un orificio uréteral. La mucosa
intestinal expuesta que yace entre las hemivejigas representa
histológicamente el área ileocecal y puede contener
hasta cuatro orificios.
El orificio más superior representa el intestino
proximal y se puede prolapsar hacia fuera y aparecer como una
deformidad en trompa de elefante, mientras que el orificio
más inferior se correlaciona con el intestino distal. El
orificio de este último representa el intestino posterior,
que termina en un saco ciego y se vincula con un ano imperforado.
Uno o dos orificios apendiculares se encuentran entre los
orificios de los intestinos proximal y distal.
En todos los casos se reconocen anormalidades
genitales. En los varones se presentan testículos no
descendidos, el pene es por lo regular bífido y cada
mitad es epispádica y está fijada a ramas púbicas
ampliamente separadas. En las mujeres, el clítoris está
dividido y habitualmente acompaña a una vagina doble y
útero bicorne.
Embriología
Típicamente, después de las 4 semanas de vida
el tabique urorectal divide la cloaca en un seno urogenital
anterior y un canal anorectal posterior.
Simultáneamente, la membrana cloacal es invadida por
pliegues del mesodermo lateral alrededor de la cuarta semana de
gestación. Se postula que si la invasión
mesodérmica no se produce, la membrana cloacal
infraumbilical persiste, con el consiguiente déficit de
desarrollo de la pared abdominal. Debido a su inestabilidad
intrínseca la membrana cloacal finalmente se rompe. Si
esto sucede antes de que descienda el tabique urorectal
entre las 6 y las 8 semanas de gestación se produce la
extrofia cloacal.
Sin embargo, en el embrión en desarrollo no existe
un estadio similar en apariencia al de la extrofia cloacal. Por
lo tanto, la anomalía no debe de representar una
detención del desarrollo embriológico sino más
bien alguna forma de defecto embriogénico. De acuerdo con
Muecke (1964), una membrana cloacal anormalmente extensa genera
un efecto de cuña que actúa como una barrera
mecánica contra la migración del mesodermo, lo que
determina el deterioro del desarrolle de la pared abdominal,
fallas en la fusión de los tubérculos genitales y
diastasis púbica. La extrofia de la cloaca se produce cuando
el efecto de cuña tiene lugar antes de la formación del
tabique urorectal, en la sexta semana.

Dibujo esquemático que
muestra los defectos urinarios y gastrointestinales en niños
de ambos sexos nacidos con extrofia cloacal. (Cortesía del
doctor Richard Hurwitz.)
Se han creado modelos de extrofia cloacal en
embriones de pollo a través de la colocación de
una cuña de plástico en el área de la membrana
cloacal para impedir la migración del mesodermo sobre la
membrana o mediante el uso de láser de dióxido de
carbono para abrir la membrana
cloacal antes de su fusión con el tabique urorectal
(Thomallia y col., 1985). Toda la evidencia experimental de
la teoría actual de la embriogénesis se basa en la
ocurrencia de este evento desencadenante en las primeras 6
a 8 semanas del desarrollo embriológico.
Sin embargo, la evidencia ecográfica de la ruptura
tardía (entre las 18 y las 24 semanas de
gestación) de la membrana cloacal, unida a informes previos de ruptura de
la membrana entre las semanas 22 y 26, arrojan dudas sobre
estas teorías. Langer (1992) y Bruch (1996) y col.,
informaron el diagnóstico prenatal de extrofia cloacal con
una membrana cloacal intacta después de 18 a 24 semanas de
gestación. Vermeij-Keers y col. (1996) postularon que
el mal desarrollo embriológico de la extrofia cloacal es
causado por una proliferación celular y una apoptosis mal
orquestados, con el resultado de una escasa formación
de mesodermo desde la placa ectodérmica umbilical. En lugar
de debatir acerca del momento exacto de la
desintegración de la membrana cloacal (8 semanas
contra 22 semanas) estos autores describieron el tabique
urorectal, la pared abdominal y la membrana cloacal como
relacionados con un origen similar (es decir, la placa
ectodérmica) y el momento de la ruptura como la
representación del balance entre la proliferación
célula la muerte celular por
apoptosis.
Anormalidades
acompañantes
Aparte de los hallazgos anatómicos comunes de la
extrofia cloacal, hasta en 85% se presentan anormalidades de
otros sistemas orgánicos distantes
al defecto básico.
Son comunes las anomalías de las vías
urinarias superiores, consistentes en riñón
pélvico, hidronefrosis, hidrouréter, atresia ureteral,
agenesia renal unilateral, riñón multiquístico,
duplicación; ureteral y ectopia fusionada cruzada. Al
revisar varias series grandes, se advirtió que tales
anormalidades se presentaban en 42 a 60% de los casos.
Se señalan defectos vertebrales en 48 a 75% de los
pacientes. Se observa mielodisplasia (mielomeningocele,
meningocele, lipomeningocele) en 29 a 46% de los sujetos. Un
informe reciente identificó
la presencia de anomalías vertebrales y medulares en 16 de
17 pacientes que se sometieron a una evaluación
completa. Por lo tanto, el índice de sospecha de esta
relación debe ser alto. Los trastornos del sistema nervioso central
distintos de la mielodisplasia son relativamente
infrecuentes. Los sobrevivientes tienen un nivel normal de
inteligencia.
Las malformaciones digestivas adjuntas consisten en
malrotación, atresia duodenal, duplicación,
divertículo de Meckel, intestino corto y apéndice
ausente o doble. En un informe, tales hallazgos estaban
presentes en 12 de 26 lactantes (46%).,0 Es importante reconocer
la presencia de un intestino delgado proximal acortado, ya
que la conservación intestinal es uno de los
principios más importantes en el tratamiento de este
trastorno. El problema puede ser
más funcional que real.Uno de los cuatro individuos
originales de Rickham murió como resultado del síndrome
de intestino corto después de sobrevivir durante dos
meses.
Muchas otras series incluyen muertes secundarias a este
síndrome. (Campbell, 2004)
También se observan defectos de las extremidades
inferiores, que consisten en pies deformes, luxación
congénita de la cadera, agenesia y otras deformidades
graves. Estas características se presentan en 26 a 30% de
los pacientes. (Campbell, 2004)
Los genitales externos en niños con extrofia
cloacal son invariablemente anormales. En los lactantes varones
el pene y el escroto pueden estar ausentes, bífidos o
epispádicos. Los testículos están uniformemente
indescendidos. El conducto deferente puede estar ausente o
ser doble. En las mujeres, el clítoris es bífido o no
existe, en tanto que la vagina puede estar ausente o ser
única, doble o extrófica. Debido a la falta de
fusión en la línea media de las estructuras de M-ller
en la mujer, casi siempre está
presente un útero bífido o duplicado. (Campbell,
2004)
CONCLUSIONES
Estudios recientes dan a conocer que la división de
la cloaca no se da por una sola masa de mesenquima, el tabique
urorectal, sino que este se compone de otras estructuras: los
pliegue de Tourneux superior y los dos pliegues de Rathke
inferolaterales.
Las anomalías de la división de la cloaca se
dan principalmente por la falta de desarrollo o de fusión de
estos pliegues, así como su fusión con la membrana
cloacal.
Las anomalías anorectales representan la
mayoría de la anomalías de la división anormal de
la cloaca por el tabique anorectal. Estas anomalías generan
fistulas (comunicaciones anormales) entre distintas partes que
establecen comunicación con el recto y con el ano, como las
fistulas rectovesicales, rectouretrales, etc.
Los términos bajo, intermedio y alto, para designar
a las malformaciones, son bastante arbitrarios y no son
útiles para fines pronósticos ni terapéuticos, por
lo que una mejor clasificación seria de acuerdo al sexo y a
la necesidad de una colostomía.
REFERENCIAS
BIBLIOGRÁFICAS
LIBROS EN
INGLES
· Muecke EC. The role of the cloacal membrane in
exstrophy : The first successful experimental study. J Urol 1964;
92 : 659.
· Gearhart JP, Jeffs RD : Campbell's Urology in The
bladder exstrophy-epispadias complex [Walsh PC et al (edrs)]. 7th
Edn; WB Saunders, Philadelphia, London, 1998, p1939
· Hurwitz RS, Manzoni GA, Ransley PG, Stephen FD.
Cloacal exstrophy : A report of 34 cases. J UroI 1987; 138 :
1060.
· Nievelstein R A J; Vermeij-Keers Chr. Anal and
ano-urogenital malformations: a histopathological study of
"imperforate anus" with a reconstruction of the pathogenesis.
1996
LIBROS EN ESPAÑOL
· Keith W. Aschcraft, MD. Cirugía
Pediátrica. 3º edición. Mc Graw-Hill
Interamericana Editores, S.A. México, D.F, 2002.
· Walsh Patrick C. Urología de Campbell.
8º edición. Editorial Médica Panamericana.
Buenos aires, 2004.
· T.W. Sadler, PhD. Embriología Médica.
10º edición. Editorial Médica Panamericana.
Madrid, España,
2006.
· Keith L. Moore. Embriología Clínica, el
desarrollo del ser humano. 7º edición. Editorial
Elsevier. Madrid, España, 2004.
LIBROS EN
LA WEB
· William D. Larsen: Embriología Humana.
3º edición. 2003. Disponible en: .
Visitada el 10 de julio del 2008.
· VV Staff, Jesús, González-Merlo, E.
González Bosquet: Ginecología. 8º edición.
2003. Disponible en:
http://books.google.com.pe/books?id=ndsTJEuDk9kC&printsec=frontcover&dq=division+cloaca&source=gbs_summary_r&cad=0.
Visitada el 10 de julio del 2008.
· Anil K. Rustgi: Requisitos de la
Gastroenterología, intestino grueso y delgado.2º
Volumen. 2005. Disponible
en:
http://books.google.com.pe/books?id=5sNuj6XhC5oC&printsec=frontcover&dq=MEMBRANA+CLOACAL&source=gbs_summary_r&cad=0.
Visitada el 10 de julio del 2008.
Autor:
Claudia Mercedes Otiniano
 Página anterior Página anterior | Volver al principio del trabajo | Página siguiente  |